
[A0001]
![]()
a) Univ. Prof. Dr. Wolfgang STADLBAUER
Institute of Organic Chemistry - Karl-Franzens University of Graz Heinrichstrasse 28,
A-8010 Graz, Austria
Tel: +43 (0)316 380 5334 Fax: +43 (0)316 380 9840
Homepage: http://www-ang.kfunigraz.ac.at/~stadlbau/
E-mail:
[email protected]
b) University of Medicine and Pharmacy, Ho-Chi-Minh City, Vietnam
Received: 15 July 1999 / Uploaded: 6 August 1999
Contents:
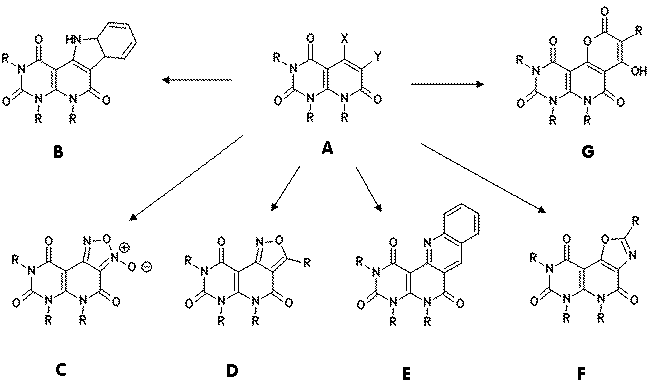
Pyrido[2,3-d]pyrimidine systems A are of chemical and chemotherapeutical
interest because they show both the properties of a 4-hydroxy-2-pyridone (present in a
series of natural products and biologically active structures, e.g. [1])
and of uracil in one ring system.
For synthetic purposes, substituents in position 5 and 6 give reactive synthons for
cyclization reactions to polyheterocyclic systems.
5-Azido-6-phenyl derivatives A (X= N3, Y = Ph) cyclize thermally to
indoles B, 5-azido-6-nitro compounds A (X= N3, Y = NO2)
react to furoxanes C, from 5-azido-6-acyl compounds A (X=N3,
Y=COR) isoxazoles D are obtained. Thermally induced cyclization to tetracyclic
quinolines E is achieved with 5-phenylamino-6-formyl compounds A (X= NHPh,
Y=CH=O). Reaction of 5-hydroxy-6-amino derivatives A (X= OH, Y= NH2)
with acyl halides gives oxazoles F. 5-Hydroxy compounds A (X=OH, Y= H) react
with malonates to give pyranes G.
 |
Determination of the suitable reaction conditions was investigated by DSC (differential
scannig calorimetry).[2]). As second aspect the reaction enthalpies
were studied for safety purposes.
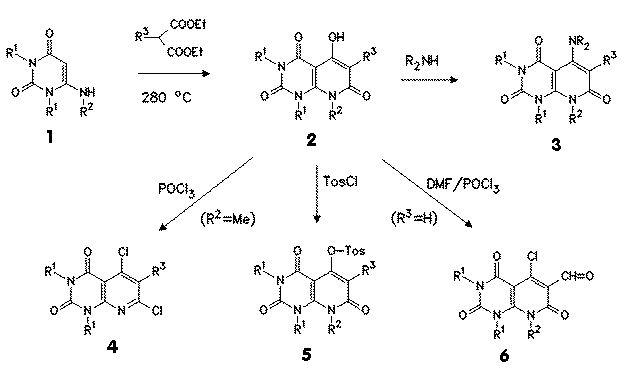
Known syntheses of pyrido[2,3-d]pyrimidine systems are described starting from 5-cyanoethyl-6-aminouracils [3] or from substituted malonic acids, acetic anhydride and 6-glucopyranosylaminopyrimidine-4-ones [4]. We have developed a simple and efficient route to 5-hydroxy-1,3,6,8-tetrasubstituted pyrido[2,3-d]pyrimidine-2,4,7-diones 2 by thermal reaction of 6-alkyl- or arylaminouracils 1 and malonates at 250oC [5].
The reactive intermediates, which were used for the following reactions as been shown in the scheme below, were obtained by reaction of 2 with amines to yield 5-amino compounds 3, and with phosphoryl chloride to obtain 5,7-dichloro compounds 4 (the methyl substituent in position 8 was cleaved during this reaction); reaction of 2 with tosyl chloride gave 5-tosyloxy-pyrido[2,3-d]pyrimidinetriones 5, and Vilsmeier reaction with DMF/phosphoryl chloride gave 5-chloro-6-aldehydes 6 from 6-unsubstituted derivatives of 2 (R3=H).
 |
Experimental example:
5-Chloro-6-formyl-1,3-dimethyl-8-phenyl-pyrido[2,3-d]pyrimidine-2,4,7(1H,3H,8H)-trione
(6, R1=Me, R2=Ph):
Phosphoryl chloride (22 mmol) was added to a cold, stirred suspension of
5-hydroxy-1,3-dimethyl-8-phenylpyrido[2,3-d]pyrimidine-2,4,7-trione (2, R1=Me,
R2=Ph) (5 mmol) in DMF (30 ml), then the reaction mixture was stirred at 50-60 oC
for 2 h. After cooling, the reaction mixture was poured into ice/water (200 ml). The
obtained yellow precipitate was filtered, washed with water and dried. Yield: 75%,
colorless prisms, mp 236-38 oC.
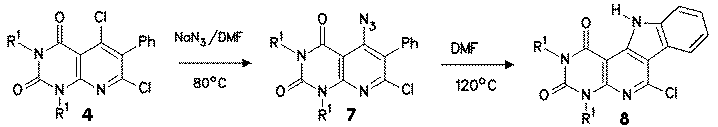
The synthesis of ortho-phenylazides 7 was performed by reaction of dichloro compound 4 by reaction with sodium azide. At room temperature in DMF only partial exchange of the 5-chloro substituent was achieved, but at 80 oC satisfactory reaction to 7 took place. The second chloro substituent in position 7 did not react with azide anion to give 7- azido- or 5,7-diazido compounds.
 |
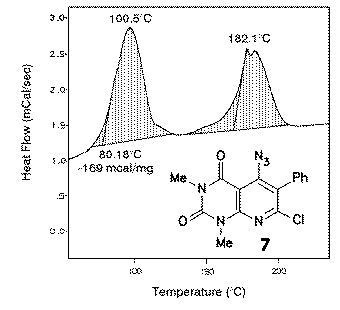 |
Thermoanalytical decomposition studies of 7 with the aid of DSC revealed that already above 80 oC a reaction took place. In synthetic experiments, best results were obtained in DMF as solvent using a temperature of about 100 - 120 oC; in this manner indolo[2,3:4,5]pyrido[2,3-d]pyrimidines 8 were obtained. The reaction enthalpy was ranging between - 150 and -190 mcal/mg, an average value compared with reactive compounds described below. |
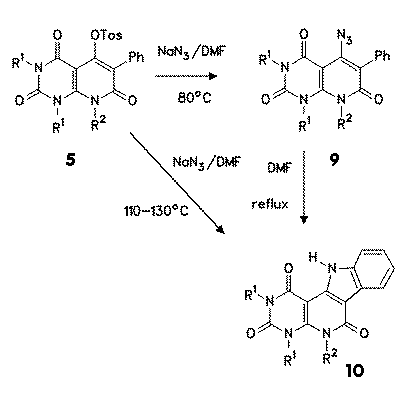 |
In a similar manner 5-tosyloxy compounds 5 reacted with sodium azide at 80 oC to 5-azido-6-phenyl derivative 9. In this case, however, no pure azide 9 could be isolated, because always already cyclized indolo derivative 10 was formed. Lower temperatures and prolonged reaction times gave lower purity and bad yields. |
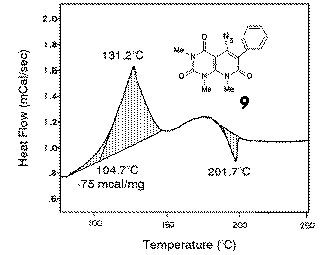 |
DSC studies of azides 9 showed a diagram which was similar to the plot obtained
for azide 7 with a peak maximum at 130 oC and an onset at 105 o.
A hint why in this case already at lower temperatures cyclization took place, can be
obtained from the base line which was not horizontally. This fact was also the reason why
no reliable valued for the reaction enthalpy could be obtained. In synthetic experiments, the thermolysis of impure azides 9 in boiling DMF gave in good yields indoles 10. Better preparative results were obtained when the tosylate 5 was reacted with sodium azide in a one pot reaction in DMF at 110-130 oC to give indoles 10. |
Experimental example: 2,4,5-Trimethylindolo[2,3:4,5]pyrido[2,3-d]pyrimidine-1,3,6-trione
(9, R1= R2= R3=Me); one pot synthesis :
A suspension of tosyloxy compound 5 (10 mmol) and sodium azide (15 mmol) in dry DMF
(30 ml) was stirred at 80-90 oC for 30 min and then at 110-130 oC
for 1 h. Then the reaction mixture was poured into ice/water (500 ml), the precipitate
filtered, washed with water and dried. Yield: 80%, mp 328-331 oC (DMF).
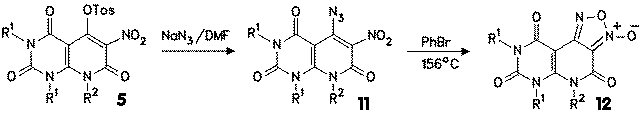
5-Azido-6-nitro compounds 11 were obtained from the 4-hydroxy compounds 2 (R3 = H) by nitration with nitric acid at room temperature (catalyzed with sodium nitrite), followed by tosylation to tosylate 5 and exchange of the tosyloxy group against the azido group with sodium azide in DMF at room temperature.
 |
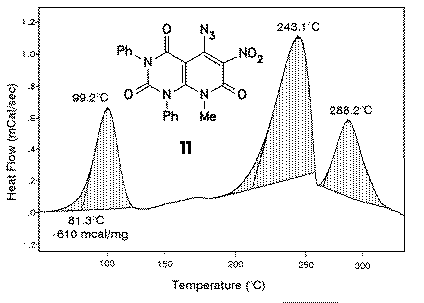 |
DSC investigation of 11 revealed that already at moderate temperatures of about 80-100 oC cyclization reaction takes place. The reaction enthalpy was found in this case to range between 500 and 700 mcal/mg, which is a strong hint for cautious handling of 11 in preparative amounts. In synthetic experiments, bromobenzene was used as the solvent which gave in excellent yields the furoxanes 12. |
Experimental example:
5-Methyl-4,7,9-trioxo-6,8-diphenyl-4,5,6,7,8,9-hexahydro-1,2,5-oxadiazolo
[3,4:4,5]pyrido[2,3-d]pyrimidine-3-oxide (12, R1=Ph, R2=
Me):
A solution of the corresponding azide 11 (10 mmol) in bromobenzene (50 ml) was
refluxed until the evolution of nitrogen had stopped (about 30 min). Then the solvent was
removed under reduced pressure and the remaining solid digested with cyclohexane (40 ml).
The product was filtered and washed with cyclohexane. Yield: 92%, yellow prisms, mp 243 oC
(toluene/cyclohexane).
 |
Desoxygenation of furoxanes was reported to lead with triphenylphosphane in good yields to oxadiazoles [6]. The temperatures reported in the literature (50-100 oC did not give the desired reactions of furoxanes 12 to oxadiazoles 13. |
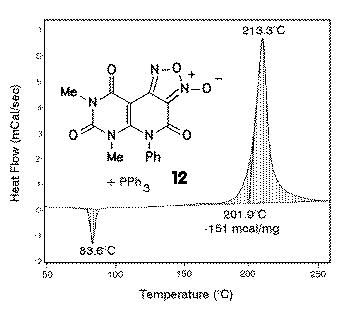 |
DSC analysis was used here in a rather unsusual way by performing the reaction of a mixture of furoxane 12 and triphenylphosphane in DSC crucibles. The DSC plot showed in this case after the melting point of triphenylphosphane (at 83.6 oC) that the desired reaction did not start below 200 oC. In a synthetic experiment, furoxane 12 was desoxygenated by triphenylphosphane at 210 oC in good yields using diphenylether as the solvent. |
Experimental example:
6,8-Dimethyl-5-phenyl-4,7,9-trioxo-4,5,6,7,8,9-hexahydro-1,2,5-
oxadiazolo[3,4:4,5]pyrido[2,3-d]pyrimidine-4,7,9(5H,6H,8H)-trione (13, R1=Me,
R2= Ph):
A mixture of the corresponding furoxane 12 (10 mmol) and triphenylphosphane 12
mmol) in 1,2-dichlorobenzene (50 ml) was heated under reflux for 8 h. After cooling the
precipitate was filtered and washed with cyclohexane to remove triphenylphosphinoxide.
Yield: 70%, yellow prisms, mp 290 oC.
 |
5-Azido-6-formyl compounds 14 which were obtained from the chloro aldehydes 6 by nucleophilic azide exchange, should give isoxazoles 15 by thermal decomposition. |
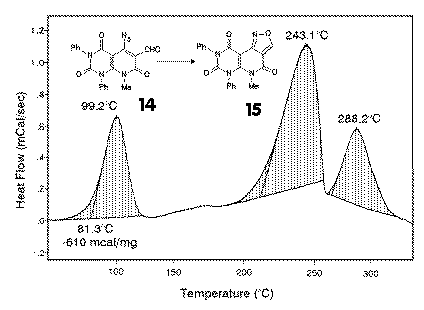 |
DSC investigation revealed, that an exothermic reaction started already at about 70-80
oC, followed by decomposition reactions at about 200 and 250 oC.
Some derivatives decomposed already at 150 oC. In a synthetic experiment, short
heating of 14 in refluxing DMF at 155 oC gave in excellent yields
isoxazolo[3,4:4,5]pyrido[2,3-d]pyrimidinetriones 15. Lower reaction
temperatures gave lower yields and impure products. For preparative purposes, a one pot
reaction starting directly from the chloro aldehyde 6 and sodium azide in DMF at
80-90 oC gave also excellent yields. The rather high reaction enthalpy of -600 mcal/mg gave again hints for safety purposes. |
Experimental example: 5,6,8-Trimethylisoxazolo[3,4:4,5]pyrido[2,3-d]pyrimidine-4,7,9(5H,6H,8H)-trione
(15 , R1=R2=Me):
A solution of
5-azido-6-formyl-1,3,8-trimethyl-pyrido[2,3-d]pyrimidine-2,4,7(1H,3H,8H)-trione 14
(10 mmol) in DMF (20 ml) was heated under reflux for 15 min. The reaction mixture was
allowed to cool and the precipitate was filtered and washed with ethanol. Yield: 96%,
colorless prisms, mp 275-277 oC (DMF).
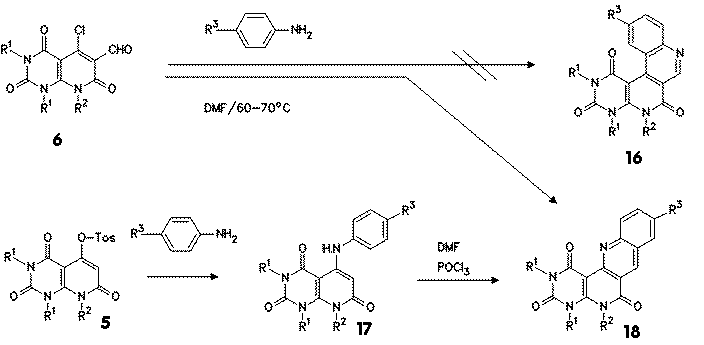
Reaction of 5-chloro-6-formyl compounds 6 with aromatic amines gave at moderate temperatures a cyclization product, which was found by unambigous synthesis to be the 1,6-naphthyridine derivatives 18, formed by a primary nucleophilic attack of the aniline at the 5-chloro substituent, followed by electrophilic attack of the aldehyde group at the aniline arene part. The same compounds, benzo[b]pyrimido[4,5-h]1,6-naphthyridine-1,3,6-triones 18 were obtained by amination of 5-tosyloxy compound 5 to the amino compound 17 followed by Vilsmeier formylation with DMF/phosphorylchloride to an intermediate aldehyde, which cyclized at the reaction conditions to give again naphthyridine 18.
 |
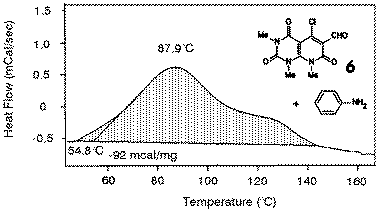 |
The most suitable reaction conditions for the cyclization of 6 to 18 were studied by DSC investigation. When the mixture of the chloro aldehyde 6 and aniline was thermolyzed, a broad exothermic signal was observed, which revealed that the reaction took place with an onset of about 55 o and a peak maximum of about 90 oC. This DSC plot shows that also such a reaction can be studied with good results by DSC. |
Experimental example:
2,4-Dimethyl-5-phenyl-9-trifluoromethoxy-benzo[b]pyrimido[4,5-h]1,6-naphthyridine-
1,3,6(2H,4H,5H)-trione (18 , R=Me, R2=Ph, R3=OCF3):
A solution of the corresponding 5-chloro-6-formyl-pyrido[2,3-d]pyrimidine-2,4,7-trione
6 (5 mmol) in DMF (30 ml) and 4-trifluoromethoxyaniline (6 mmol) was stirred at
60-70 oC for 2 h. After this time a yellow precipitate was formed which was
filtered after cooling, washed with acetone, dried and recrystallized from DMF. This
product could be shown to be the hydrochloride of 18 which gave upon treatment with
aqueous 2 N sodium hydroxide solution (50 ml) the free base 18. Yield: 80%, mp
263-265 oC from DMF/ethanol.
5-Hydroxy-6-nitro derivatives 19, which were obtained from 5-hydroxy compounds by nitration with nitric acid/sodium nitrite in acetic acid at room temperature, cyclized to 2-alkyl-oxazolo[5,4:4,5]pyrido[2,3-d]pyrimidinetriones 20 by reduction with zinc in the presence of alkanoates. Ring opening of the oxazole ring with hydrochloric acid gave the 6-amino hydrochlorides 21, which in turn could be cyclized again with benzoic acid chlorides in the presence of polyphosphoric acid to give 3-aryl-oxazolo[5,4:4,5]pyrido[2,3-d]pyrimidinetriones 22.
 |
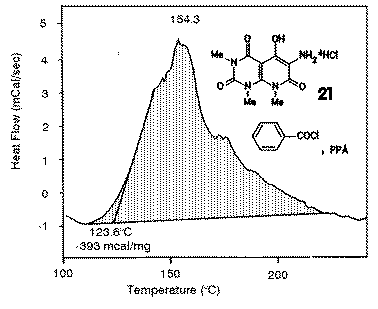 |
The reaction conditions of the cyclization of 21 to 22 were studied by DSC in order to obtain the suitable reaction temperatures. The diagrams showed an exothermic reaction with an onset of about 120 oC and a peak maximum of about 150 oC. In preparative experiments, the reaction was performed at this temperature. |
Experimental example:
2-(4-Nitrophenyl)-5,6,8-trimethyloxazolo[5,4:4,5]pyrido[2,3-d]pyrimidine-4,7,9(5H,6H,8H)-trione
(22 , R1=R2=Me, Ar=4-NO2-Ph):
A mixture of the corresponding 6-amino-5-hydroxy-pyrido[2,3-d]pyrimidine-2,4,7(1H,3H,8H)-trione
hydrochloride 21 (10 mmol) with 4-nitrobenzoylchloride (30 mmol) in polyphosphoric
acid (20 g) was heated to 150 oC for 4 h. The warm reaction mixture was poured
into ice/water (400 ml), brought to pH 6-7 with aqueous concentrated sodium hydroxide
solution. The formed precipitate was filtered by suction, washed with water, dried and
recrystallized from the appropriate solvent. Yield: 71%, mp 350 oC.
5-Hydroxy compounds 2 having no substituent in position 5 afforded in a 1:1 condensation reaction with monosubstituted diethylmalonates in boiling diphenylether pyrano[3,2:4,5]pyrido[2,3-d]pyrimidinetetraones 23. With unsubstituted malonates no cyclization products were isolated. Best results were obtained with phenylmalonates, but also alkylmalonates such as ethyl-, butyl-, benzyl- or allylmalonates gave in good yields the corresponding pyranes.
As intermediate the formation of a ketene is assumed. This assumption is supported by ir measurements. Attempts to study this reaction with DSC failed, however, because no distinguished reaction peak could be observed.
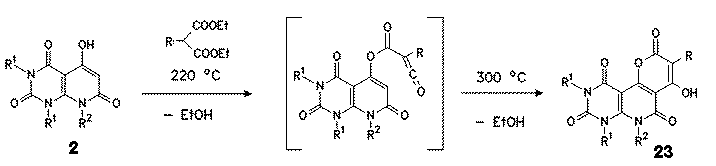 |
Experimental example:
4-Hydroxy-6,7,9-trimethyl-pyrano[3,2:4,5]pyrido[2,3-d]pyrimidine-2,5,8,10(6H,7H,9H)-tetraone
(23 , R1=R2=Me, R = Ph):
A mixture of 5-hydroxy-1,3,8-trimethyl-pyrido[2,3-d]pyrimidine-2,4,7-trione 2
(10 mmol) and diethyl phenylmalonate (20 mmol) in diphenylether (15 ml) was heated for 2 h
to 250-260 oC using a short air condenser to remove liberated ethanol. After
cooling, the reaction mixture was digested with cyclohexane (50 ml), the obtained
precipitate was filtered, washed with cold cyclohexane and dried. Then the precipitate was
boiled with ethanol for 10 min, cooled, filtered by suction, washed with ethanol and
recrystallized from DMF. Yield: 73%, yellow prisms, mp 310-312 oC (DMF).
Most of these compounds have been tested in antitumor and anti-AIDS programs of the US-NCI, in animal health programs, and in a program for anti tuberculosis and antimicrobial activity (TAACF) by the US National Institute of Allergy and Infectous Diseases. Some compounds have shown interesting results in in vitro tests, however in vivo tests have not confirmed the activities.
This work was supported by the FWF (Osterreichischer Fonds zur Forderung der wissenschaftlichen Forschung) project No. P 10785-CHE .
[1] The Merck Index, 12th Edition, S. Budavari, ed., Merck, Sharp & Dohme, Rahway,
NJ, 1996, 6308 (Mocimycin), 8377 (Ricinine); M. J. Robins, C. Kaneko, M. Kaneko, Can. J.
Chem. 1981, 59, 3356; H. J. Knops, L. Eue, R. R. Schmidt, Bayer AG, German Offen. DE 3 210
598 (1983); Chem. Abstr. 1984, 100, 4412j; The Merck Index, 12th Edition, S. Budavari,
ed., Merck, Sharp & Dohme, Rahway, NJ, 1996, 7657 (Piroctone).
[2] All DSC data were obtained on a Rheometric Scientific
DSC-Plus instrument with the DSC software V 5.42. The DSC plots were recorded between
25-500oC, the heating rate was 2-10oC/min, using 1.5-3 mg of
substance in sealed aluminium crucibles (11 bar).
[3] O. A. Burova, N. M. Smirnova, V. I. Pol'shakov, G. S. Chernov, G. A. Losev, T. S.
Safonova, Khim. Geterotsikl. Soedin. 1991, 674
[4] L. Quijano, M. Nogueras, M. Melgarejo, A. Sanchez, Monatsh. Chem. 122, 255
(1991).
[5] A.F.A. Khattab, Dang V. T., W. Stadlbauer, J. Prakt. Chem. 338, 151 (1996).
[6] F. Yoneda, Y. Sakuma, J. Heterocycl. Chem. 10, 993 (1979); E. C. Taylor, Y.
Maki, A. McKillop, J. Org. Chem. 37, 1601 (1972).
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0001 as the message subject of your e-mail.{kind=link}