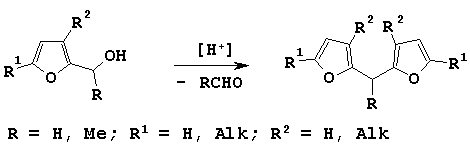
[A0005]
SYMMETRIC DIFURYL- AND TRIFURYLMETHANES:
GENERAL METHODS OF SYNTHESIS
Tat'yana A. Stroganova, Alexander V. Butin, Lyudmila N. Sorotskaya, Vladimir G. Kul'nevich
Research Laboratory of Furan Chemistry, Kuban State Technological University, Moskovskaya 2, Krasnodar, 350072 Russia
E-mail:
[email protected], [email protected]Received: 30 July 1997 / Uploaded: 6 August 1997
INTRODUCTION
Although at last time the most of investigators put their attention to development of convenient approaches to synthesis of asymmetric furylhetarylalkanes, symmetric di- and trifurylmethanes keep their importance as before. Some derivatives of difurylalkanes use in various fields of industry.
Our stydies let us to elaborate convenient routes to several derivatives of symmetric di- and trifurylmethanes series.
DIFURYLALKANES
The self-condensation of furfurylic alcohols is of most interest among methods of synthesis of symmetric difurylmethanes.
Difurylmethane was isolated from mixture of products resulted from oligomerization of furfurylic alcohol in acidic condition [1]. It was found that diamine of difurylmethane series as a minor product has been obtained on reaction of 5-hydroxymethylfurfurylamine with 5.1 M HCl [2]. Hydrothermolysis of alkylfurylcarbinoles at 300?C and pH~7 [3] produced the corresponding difurylalkanes in 15 mole % yield.
In all above-listed papers the preparing of difurylalkane represents the by-reaction, which due to low yields of latter is not important for synthetic aims.
At the same time, there were proposed preparative approaches to synthesis of difurylalkanes from 3,5-dialkylsubstituted 2-furylcarbinoles in excellent yields. These reactions proceeded in the presence of polyphosphoric acid [4], Ag (I) ions or Cl3CCOOH [5].
For the first time we faced the self-condensation of furfurylic alcohols reducing 5-methylfurfural and acetylsylvan with sodium borohydride in ethanol. Difurylmethanes were fixed under acidification of the reaction mixture with diluted solution of hydrochloric acid.
Continuing our investigation on conversion of furylcarbinoles in acidic conditions we have tried to develop a simple and efficient procedure for preparing of symmetric difurylalkanes.
In literature we found data [6] devoted to tuberculostatic activity of some representatives of bis(5-arylfuryl-2)methanes that up to date were obtained only as by-products of some reactions.
Therefore, we have chosen 5-arylfurfurylic alcohols as initial compounds.
We have discovered that these alcohols transformed into difurylmethanes 2 in the presence of HClO4 in dioxane at room temperature.
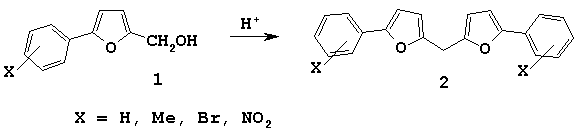
Interaction of compounds 1 with sylvan in similar conditions caused the formation of mixture of symmetric 2 and asymmetric 3 products. It should be noted that in the most cases there were formed mainly symmetric difurylmethane 2. Analogous conversions were described earlier by Balaban and co-workers [4] for furan derivatives and Jackson et al. for 2-hydroxymethylene pyrroles [7].

There were offered two possible pathways for the formation of difurylalkane products [4, 5]:
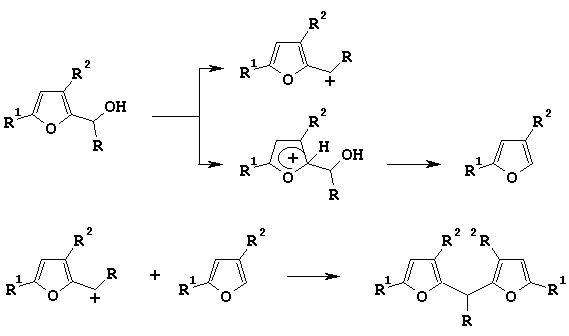
Scheme 1
Scheme 2
However, we could find no information on the behavior and transformation of furylarylmethanoles in the acid conditions.
We have treated (5-methylfuryl-2)arylcarbinoles 4 with various acid catalysts but we could not detect any corresponding difurylmethanes. This fact is agreed with above-mentioned mechanisms.
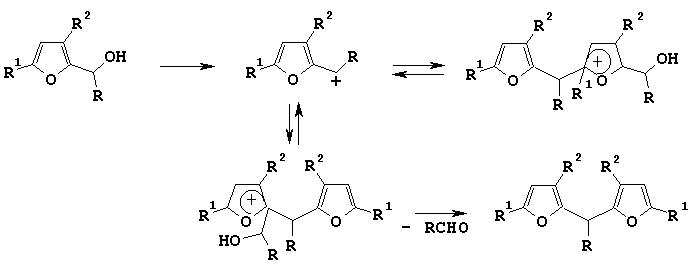
According to them there are two directions for electrophilic attack on the molecule of alcohol - on oxygen of hydroxy-group and on furan ring - and as a result two cations can be prepared. In the case of furfurylic alcohols and 1-furylethanoles generated cations are similar in their stability, and it is favorable for difurylmethane producing.
For furylarylcarbinoles 4 the generation of cation A (Scheme 3) is more preferable because it is the most stable of two cations which can be formed during the electrophilic attack.
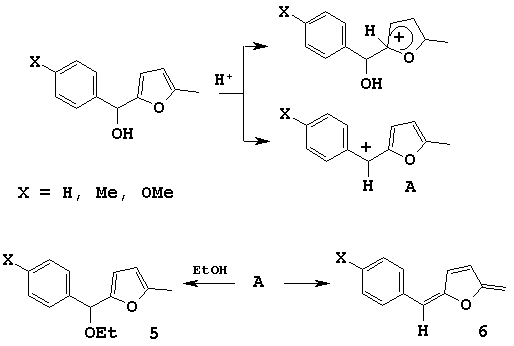
Scheme 3
When the reaction proceeded in ethanol this cation A rearranged into ether 5 with excellent yield (about 80 %). Under treating of furylarylmethanols with acid catalysts in such solvents as benzene and dioxane we observed the strong tarification of reaction mixture that could be caused by dihydrofuran structure 6 formation.
Now our study is in progress.
TRIS(5-R-FURYL)METHANES
The syntheses of symmetric trifurylmethanes have not been the objective of so many publications as difurylmethanes.
The most familiar methods of their syntheses are condensation of furfurals with corresponding furan substrates or interaction between furan compounds and CHCl3 [8]. We found the formation of trifurylmethane under treatment of 2-(5-methylfuryl-2-)-1,3-dioxolane with trityl perchlorate. It should be mentioned that this reaction proceeded in unusual path, and instead of desired dioxolanium salt we isolated trifurylcarbenium perchlorate.
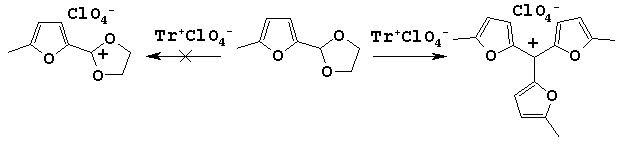
Trifurylmethanes was found as a minor product by GLC-method. Its formation, in our opinion, was caused by furyldioxolane transformation under the action of electrophilic reagent (acid).
Analogous conversion was described by Clezy et al [9]. They discovered that in the interaction of pyrrolic aldehyde with ethylene glycol in the presence of p-TsOH as a catalyst the product was tripyrrylmethane instead of an expected acetal.

Taking into account our results and literature data we set ourselves the task of developing convenient and simple route to symmetric tris(5-R-furyl)methanes.
For this purpose we studied the influence of acidic catalysts on the direction and selectivity of the reaction of 5-methylfurfural and ethylene glycol.
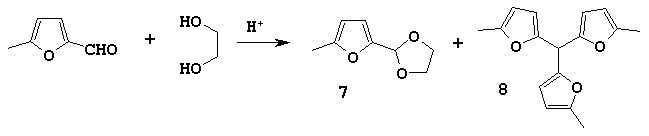
The obtained results are listed in the Table.
Table
Catalyst |
Ratio 7 : 8 (GLC data) |
KU-2 (10 % of 5-methylfurfural mass) |
7, traces of 8 |
Amberlyst 15 (10 % of 5-methylfurfural mass) |
1 : 20 |
Amberlyst 15 (50 % of 5-methylfurfural mass) |
8, traces of 7 |
p-TsOH |
1 : 3 |
HClO4 |
5 : 1 |
Et2O BF3 |
8, traces of 7 |
We found that the application of weak acidic catalyst such as an ion-exchanged resin KU-2 allowed preparing of 2-(5-methylfuryl-2)-1,3-dioxolane 7 and, at the same time, using of stronger catalysts produced trifurylmethane 8 formation.
The ion-exchanged resin Amberlyst 15 turned out to be a universal catalyst: varying its quantity we could change the course of reaction and isolate mainly either dioxolane 7 or symmetric trifurylmethane 8.
We established that in the same reaction conditions the utilisation of 1,3-butanediol instead of ethylene glycol gave the worse results: there was obtained only the mixture of trifurylmethane and dioxane. This result conformed to Clezy's data [9].
To carefully study this reaction we have investigated another furfurals, in particular, furfural, 5-bromofurfural, 5-ethylfurfural and 5-arylfurfurals.
2-(Furyl-2)-1,3-dioxolane was found to be the main product of the interaction furfural and ethylene glycol in the presence of p-TsOH. Its yield was about 80 %. When we tried to apply stronger acid catalysts they produced the tarification of the reaction mixture due to polymerisation on 5th position of furan ring.
In the case of 5-bromfurfural we could not isolate trifurylmethane because the reaction has stopped at the 2-(5-bromfuryl-2)-1,3-dioxolane formation stage.
The condensation of 5-ethylfurfural with ethylene glycol did not differ from the reaction of methylfurfural, and as a result we obtained corresponding symmetric tris(5-ethylfuryl-2)methane in a good yield.
The proposed approach to syntheses of symmetric trifurylmethanes is very important for 5-arylfurfurals. The point is that to obtain symmetric tris(5-arylfuryl-2)methanes by the traditional methods one should have not easily accessible 2-arylfurans. At the same time, 5-arylfurfurals used in our reaction can be easily derived by the Meierween reaction.
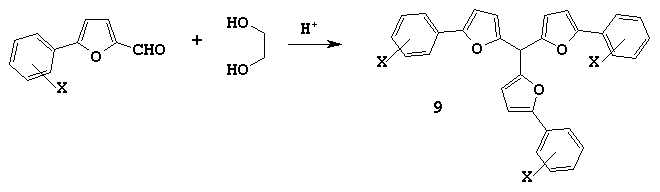
We have synthesised several symmetric tris(5-arylfuryl-2)methanes 9 based on 5-arylfurfurals:
We have discovered that the condensation of 5-(4-nitroarylfuryl-2)-furfural with ethylene glycol in benzene under boiling produced only corresponding 2-[5-(4-nitrophenyl)furyl-2]-1,3-dioxolane but tris[5-(4-nitrophenyl)furyl-2)]methane was not fixed in the reaction mixture.
The investigation is continued.
References
[1] Wewerka E.M., Loughran E.D., Walters K.L., J. Appl. Polym. Sci., 15, 1437 (1971).
[2] Holfinger M.S., Conner A.H., Holm D.R., Hill C.G. Jr., J. Org. Chem., 60, 1595 (1995).
[3] Nelson D.A.; Hallen R.T., J. Anal. Appl. Pyrolysis, 12, 11 (1987).
[4] Balaban A.T., Bota A., Zlota A., Synthesis, 2, 136 (1990).
[5] Marshall J.A., Wang X., J. Org. Chem., 56, 960 (1991).
[6] Oleynik A.F., Dozorova E.N., Solov'eva N.P., Khim.-Pharm. Zhurnal, 17, 928 (1983).
[7] Jackson A. H., Pandey R. K., Rao K. R. N., Tetrahedron Lett., 26, 793 (1985); Gonsalves A. M. d’A. R., Kenner G. W., Smith K. M., Tetrahedron Lett., 3, 2203 (1972).
[8] a) Kul'nevich V.G., Solonenko L.A., Zhuravlev S.V., Khim. Geterotsikl. Soedin., 5, 592 (1983). b) Zhuravlev S.V., Kul'nevich V.G., Khim. Geterotsikl. Soedin., 5, 597 (1983).
[9] Clezy P.S., Fookes C.J.R., Lau D.Y.K., Aust. J. Chem., 27, 357 (1974).
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0005 as the message subject of your e-mail.