Third International Electronic Conference
on Synthetic Organic Chemistry (ECSOC-3), www.mdpi.org/ecsoc-3.htm, September 1-30, 1999
[A0006]
Asymmetric Synthesis of
Quaternary a-Amino Acids
Using D-Ribonolactone Acetonide as a Chiral Auxiliary
Marcial Moreno-Mañas, Elisenda Trepat, Rosa M.
Sebastián and Adelina Vallribera*
Department of Chemistry. Universitat Autònoma
de Barcelona. Cerdanyola. 08193-Barcelona. Spain
E-mail: [email protected]
Received: 22 July 1997 / Uploaded: 5 August 1997
Abstract 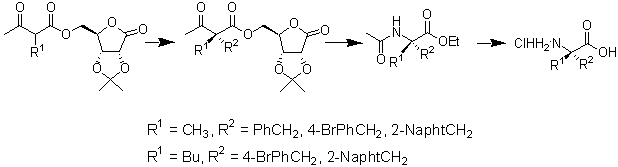
Preparation of a,a -disubstituted glycines is a
matter of current interest owing to their importance as enzyme inhibitors and peptide
modifiers. Herein, we describe a new simple methodology to prepare enantiopure a,a
-dialkylglycines based on the use of commercially available D-ribonolactone as chiral
auxiliary. Enantiopure a -methyl and a -butyl series are prepared through
diastereoselective alkylation and posterior Schmidt rearrangement of a,a
-dialkylacetoacetates of D-ribonolactone acetonide. Absolute configuration was assigned
through preparation of enantiopure 4,4-disubstituted 3-methyl-2-pyrazolin-5-ones.
The high interest in the preparation of
enantiomerically pure a,a -disubstituted
glycines is based on their remarkable properties as enzyme inhibitors [1]
and as conformational modifiers of peptides [2]. Especially a-methyl series have been extensively studied [3].
Due to the
current interest in these compounds many synthetic methodologies have already been
developed [4]. One synthetic
approach is the use of cyclic derivatives such as Schöllkopf's bis(lactim)ether, prepared
generally from L-tert-leucine and including two amino ester separation in the
process of isolation of desired products. Others cyclic substrates are Seebach's
oxazolidinones, imidazolidinones and 2,5-dihydroimidazoles [5] their synthesis including classical resolution
through diastereoisomeric salt formation or chromatographic separation on a chiral column.
Nájera's oxazinones and tetrahydropyrazinones [6] have been also used with real success. Also
remarkable are the results from Davies, who has used oxazolidinones derived from
ferrocenecarbaldehyde and sodium (S)-alaninate [7] and the work
of Sandri using chiral morpholine derivatives [8]. An alternative involves the palladium-catalyzed
asymmetric allylation of azlactones [9].
A different approach is based on the
diastereoselective alkylation of activated methylene groups in open chain compounds.
Remarkable diastereoselective alkylations have been achieved for 2-cyanoesters of
enantiopure dicyclohexylsulfamoylisoborneol by Cativiela and coworkers [10]
(1, Z=CN). After the appropriate rearrangement process the diastereomerically pure a, a-dialkylcyanoacetate can be elaborated to
afford both enantiomers of the same amino acid. An alternative consists of the
incorporation of the chiral auxiliary in the form of an enamine (2). Koga has
reported [11] the alkylation of lithioenamines derived from a-alkyl-b-ketoesters and (S)-valine tert-butyl ester. The a,a-dialkyl- b-ketoesters obtained had been used by Georg and coworkers as
intermediates to prepare a,a-disubstituted glycines [12].
Fukumoto et al. have worked on chiral derivatives of malonic acid. Precursors of amino
acids are prepared through diastereoselective alkylation of 8-phenylmenthyl a-alkylmalonic monoesters (1, Z=COOH)
followed by several transformations including a Curtius rearrangement [13].
Figure 1
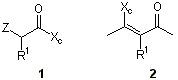
We have previously reported the preparation of
enantiopure diphenylmethyl-, 9-fluorenyl and (1-adamantyl)glycines through cobalt mediated
alkylation of (4R) and (4S)-3-acetoacetyl-4-benzyloxazolidin-2-ones (1,
Z=acetyl, R1=H) [14]. Several dialkylation attempts on the
same substrate failed. However alkylation of (-)-8-phenylmenthyl 2-methylacetoacetate
affords 8-phenylmenthyl 2-alkyl-2-methylacetoacetates in a maximal diastereomeric ratio of
85:15 [15]. Posterior elaboration to amino acids failed probably due
to steric hindrance. This previous results made us to consider other alcohols as chiral
auxiliaries. We chose D -ribonolactone for two main reasons: (a) It is a commercially
available sugar derivative (b) Being a primary alcohol, its ester might be easily
manipulated (this has failed in the case of (-)-8-phenylmenthol).
2,3-O-isopropyliden-[16] and 2,3-O-cyclohexyliden-g-D-ribonolactone [17] had been prepared by
reactions previously described in the literature. These protected lactones react with
2,2,6-trimethyl-1,3-dioxen-4-one, 5, in refluxing toluene to afford the
corresponding methyl acetoacetates, 6 and 7 (74-82%) (Scheme 1). Methylation
at C- a is carried out with potassium carbonate, methyl iodide
in acetone at 40oC (69-71%). We have first studied dialkylation process through
generation of enolate with NaH at –78oC and posterior addition of
benzyl bromide, and we obtained a dr similar for the two chiral auxiliaries. In the case
of cyclohexylidene protection we were unable to isolate the major diastereoisomer in pure
form. Therefore, we chose 2,3-O-isopropyliden- g - D
-ribonolactone as chiral auxiliary.
Scheme 1
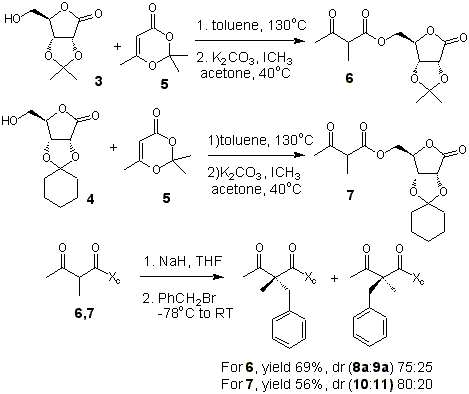
Some attempts have been made to optimize the
process: (a) Other bases such as LDA, phosphazene P4-t-Bu in n-hexane
(C22H63N13P4) and sodium
bis(trimethylsilyl)amide gave similar or worse dr; (b) Addition of N,N'-dimethylpropyleneurea
(DMPU) or change of THF to DMPU, when using NaH, did not give better results.
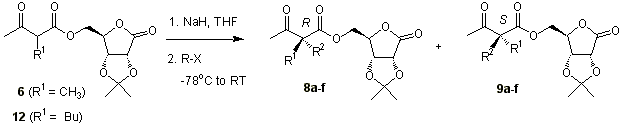
Alkylation of 6
with a series of alkyl halides furnished compounds 8a-d and 9a-d in
reasonable diastereomeric excesses (Table 1) [18]. The major
diastereoisomers were isolated in pure form in all cases except for R2 =
PhCH=CHCH2.
We were also
interested in disubstituted glycines with one group different from methyl. Compound 12
was easily prepared from 2,3-O-isopropyliden- g - D
-ribonolactone acetoacetate with NaH in refluxing THF and n-butyl iodide in 71%
yield. Even with the bulkier n-butyl substituent high yields are obtained in the
dialkylation process (Table 1).
Table 1 . Results of diastereoselective dialkylation of 6 and 12
with a series of alkyl halides
.
R1
|
R2
|
productsa[19] |
yield (%) |
dr
|
CH3
|
PhCH2
|
8a+9a
|
69
|
75:25
|
CH3
|
4-BrPhCH2
|
8b+9b
|
64
|
78:22
|
CH3
|
PhCH=CHCH2
|
8c+9c
|
71
|
80:20
|
CH3
|
2-NaphtCH2
|
8d+9d
|
74
|
80:20
|
Bu
|
4-BrPhCH2
|
8e+9e
|
75
|
80:20
|
Bu
|
2-NaphtCH2
|
8f+9f
|
69
|
80:20
|
a
All pure diastereoisomers 8a,b,d-f and 9a,b,d-f
and the mixture 8c+9c gave correct elemental analysis (C, H).
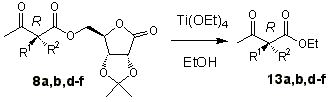
Table 2 summarizes the results for
transesterification of pure diastereoisomers 8a,b,d-f [20]. By
using an excess of titanium(IV) tetraethoxidein refluxing ethanol the corresponding
enantiopure ethyl a,a -dialkyl acetoacetates are obtained in
excellent yields when R1 = CH3. Experiments with sodium ethoxide
gave worse results. For butyl substituent as in compounds 8e,f, less efficient
reactivity is found, as expected.
Table 2. Results of
transesterification reaction.
R1
|
R2
|
productsa
|
bp
(mmHg) |
yield (%) |
CH3
|
PhCH2
|
13a[11][21] |
125 oC.
(0.07) |
92
|
CH3
|
4-BrPhCH2
|
13b
|
150
oC. (0.07) |
90
|
CH3
|
2-NaphtCH2
|
13d
|
175 oC.
(0.07) |
93
|
Bu
|
4-BrPhCH2
|
13e
|
150 oC. (0.07) |
37
|
Bu
|
2-NaphtCH2
|
13f
|
-
|
49
|
a
All products 13 gave correct elemental analysis (C, H).
Schmidt rearrangement on 13 with
sodium azide and methanesulfonic acid in dimethoxyethane afforded acetamides 14 [22]. We have previously recommended the use of DME as an alternative to
the unsafe chlorinated solvents normally used in Schmidt rearrangement [23].
Acetamides 14 were hydrolyzed in refluxing 6M HCl to give the amino acids
hydrochlorides 15 in excellent yields.
Table 3. Schmidt
reaction of a,a -disubstituted
acetoacetates 13 and posterior hydrolysis of acetamides 14.
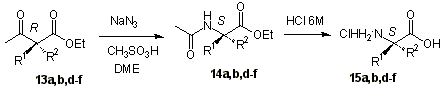
R1
|
R2
|
14
(%)a |
15(%)b
|
15,
[a]D |
CH3
|
PhCH2
|
14a[13b](82) |
15a
(81) |
-7
(c 1.22, H2O) |
CH3
|
4-BrPhCH2
|
14b
(63) |
15b
(81) |
-8
(c 1.05, H2O) |
CH3
|
2-NaphtCH2
|
14d
(82) |
15d
(76) |
5 (c
1.14, EtOH) |
Bu
|
4-BrPhCH2
|
14e
(43) |
15e
(73) |
7 (c
1.06, EtOH) |
Bu
|
2-NaphtCH2
|
14f
(41) |
15f
(65) |
-3
(c 0.90,EtOH) |
a
Compounds 14[24]gave correct elemental analysis. b Compounds
15a,b,d,e[25]gave good elemental analysis.
Compound 15f [25]
gave correct elemental analysis for C and N.
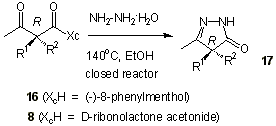
We have previously
described a method for the preparation of enantiomerically pure (4R)-4,4-disubstituted
2-pyrazolin-5-ones from (-)-8-phenylmenthyl (2R)-2-alkyl-2-methylacetoacetates, 16[26]. Reaction of 16 with hydrazine hydrate afforded (4R)-4-alkyl-3,4-dimethyl-2-pyrazolin-5-ones,
17 (Table 4). X-Ray difracction studies on 16a and 16b showed R
configuration at C-a. Accordingly, the new stereogenic center of 17a
and 17g was also R. Compounds 8a,b,d-f were converted into
17a,b,d-f in excellent yields. Compound 17a obtained from 8a has the
same [a]D as the one obtained from 16a.
Configuration to 17b,d-f was assigned by comparison of the circular dichroism with
those of 17a and 17g. All of them present a strong negative Cotton effect.
Therefore the major diastereoisomers obtained in the dialkylation process using D
-ribonolactone acetonide as chiral auxiliary have R absolute configuration at C-a, and consequently enantiopure hydrochlorides of (S)-a-alkyl alanines, 15a,b,d, and (S)- a -alkyl- a -butylglycines glycines, 15e,f,
had been prepared.
Table 4. Preparation of enantiomerically pure 4,4-disubstituted
2-pyrazolin-5-ones, 17.
| |
R1
|
R2
|
productsa
|
yield (%) |
[a]Db |
16a
|
CH3
|
PhCH2
|
17a[26] |
95
|
-186 (c 1.24) |
16b
|
CH3
|
4-ClPhCH2
|
17g[26] |
88
|
-87 (c 0.12) |
8a
|
CH3
|
PhCH2
|
17a
|
92
|
-180 (c 1.20) |
8b
|
CH3
|
4-BrPhCH2
|
17b
|
86
|
-144 (c 1.01) |
8d
|
CH3
|
2-NaphtCH2
|
17d
|
75
|
-211 (c 0.88) |
8e
|
Bu
|
4-BrPhCH2
|
17e
|
74
|
-88 (c 1.04) |
8f
|
Bu
|
2-NaphtCH2
|
17f
|
70
|
-127 (c 1.01) |
a
Compounds 17b,d,e[27]
gave correct elemental analysis. Compound 17f gave good HRMS.
b All determined in CHCl3.
In conclusion, we have developed a new simple method
for the synthesis of enantiopure (S)- a,a -disubstituted
glycines using D -ribonolactone acetonide as a chiral auxiliary. The advantages of this
approach are: (a) The facility to obtain the chiral auxiliary (only protection of
available material is needed); (b) The fact that only one diastereomeric separation is
needed (classical column chromatography); (c) The method allows the introduction of
substituents bulkier than methyl.
Acknowledgement Financial
support from DGICYT (Ministry of Education and Science of Spain; project PB93-0896 and
predoctoral grant to E.T.) and from CIRIT (Generalitat de Catalunya; project SGR98-0056
and predoctoral grant to M.R.S) is gratefully acknowledged.
References and Notes
[1] Heimgartner, H. Angew.
Chem. Int. Ed. Engl. 1991, 30, 238-264.
[2] Cheng, H.; Keitz, P.;
Jones, J. B. J. Org. Chem. 1994, 59, 7671-7676.
[3] Toniolo, C.; Formaggio,
F.; Crisma M.; Valle, G.; Boesten, W. H. J.; Schoemaker H. E.; Kamphuis, J.; Temussi, P.
A.; Becker, E. L.; Précigoux, G. Tetrahedron1993, 49, 3641-3653.
[4] (a) Cativiela,
C.; Díaz-de-Villegas, M. D. Tetrahedron: Asymmetry, 1998, 9,
3517-3599 (Report 40). (b) Duthaler, R. O. Tetrahedron1994, 50,
1539-1650 (Report 349). (c) Williams, R. M. Synthesis of Optically Active a-Amino Acids; Pergamon Press: Oxford, 1989.
(d) Wirth, T. Angew. Chem. Int. Ed. Engl 1997, 36, 225-227.
[5] Seebach, D.; Hoffman M.
Eur. J. Org. Chem. 1998, 1337-1351.
[6] (a) Chinchilla, R.;
Falvello L. R.; Galindo, N.; Nájera C. Angew. Chem. Int. Ed. Engl. 1997, 36,
995-997. (b) Abellán, T.; Nájera, C.; Sansano, J.M. Tetrahedron: Asymmetry, 1998,
9, 2211-2214.
[7] Alonso, F.; Davies, S.
G., Elend, A. S.; Haggitt, J. L. J. Chem. Soc., Perkin Trans. I, 1998,
257-264.
[8] Carloni, A.; Porzi, G.;
Sandri, S Tetrahedron: Asymmetry 1998, 9, 2987-2998.
[9] Trost, B. M.; Ariza, X.
Angew. Chem. Int. Ed. Engl. 1997, 36, 2635-2637.
[10] (a) Cativiela, C.;
Díaz-de-Villegas, M. D.; Galvez, J. A. Tetrahedron: Asymmetry 1994, 5,
261-268. (b) Cativiela, C.; Díaz-de-Villegas, M. D.; Galvez, J. A. Tetrahedron1994,
50, 9837-9846. (c) Cativiela, C.; Díaz-de-Villegas, M. D. Tetrahedron 1995,
51, 5921-5928. (d) Badorrey, R.; Cativiela, C.; Díaz-de-Villegas, M. D.; Galvez,
J. A.; Lapeña, Y. Tetrahedron: Asymmetry 1997, 8, 311-317.
[11] Ando, K.; Takemasa,
Y.; Tomioka, K.; Koga, K. Tetrahedron 1993, 49, 1579-1588.
[12] Georg, G. I.; Guan,
X.; Kant, J. Tetrahedron Lett. 1988, 4, 403-406.
[13] (a) Ihara, M.;
Takahashi, M.; Niitsuma, H.; Taniguchi, N.; Yasui, K.; Fukumoto, K. J. Org. Chem.1989,
54, 5413-5415. (b) Ihara, M.; Takahashi, M.; Nitsuma, H.; Taniguchi, N.; Yasui, K.;
Fukumoto, K. J. Chem. Soc. Perkin Trans. I, 1991, 525-535.
[14] Gálvez, N.;
Moreno-Mañas, M.; Vallribera, A.; Molins, E.; Cabrero, A. Tetrahedron Lett. 1996,
37, 6197-6200.
[15] Moreno-Mañas, M.;
Sebastián, R.M.; Vallribera, A.; Molins, E.; Espinosa, E. Tetrahedron: Asymmetry, 1997,
8, 1525-1527.
[16] Hough, L.; Jones, J.
K. N.; Mitchell, D. L. Can. J. Chem. 1958, 36, 1720-1728.
[17] Beer, D.; Meuwly, R.;
Vasella, A. Helv.. Chim. Acta 1982, 65, 2570-2582.
[18] General
Procedure for Diastereoselective Alkylation of Compounds 6 and 12. Compound 6
(4.65 g, 16.2 mmol) in anhydrous THF (12 mL) was added to magnetically stirred NaH (60%
suspension in mineral oil, 0.84 g (21.1 mmol)) in anhydrous THF (10 mL) at room
temperature and under nitrogen atmosphere. Thereaction mixture was stirred for 15 minutes,
cool down to –78oC and then a solution of benzyl bromide (4.17 g,
24.2 mmol) in anhydrous THF (10 mL) was added. The reaction was allowed to warm to room
temperature and was stirred for 12 h. THF was evaporated, and the residue partitioned
between CH2Cl2:H2O. The aqueous layer was washed with CH2Cl2
(3x20 mL). The combined dichloromethane extracts were dried with anhydrous sodium sulfate
and the solvent was evaporated to give a crude which was a mixture of 8a and 9a
in a ratio ca. 75:25 determined by integration of the 1H NMR signal of the CH3CO
protons at d 2.10 and 2.19. The
crude was chromatographed on silica gel under pressure, eluting with hexane: diethyl ether
mixtures of increasing polarity to afford 2.81 g (46%) of 8a and 1.04 g (17%) of 9a.
8a: white solid; mp 101-103oC.; [ a ]D = 16 (c = 1.09, CHCl3); IR (KBr) 1785 (s),
1729 (s), 1715 (s) cm-1; 1H NMR (250 MHz, CDCl3) d 1.23 (s, 3H), 1.29 (s, 3H), 1.37 (s, 3H),
2.11 (s, 3H), 2.90 (d, J = 13.1 Hz, 1H), 3.17 (d, J = 13.1 Hz, 1H),
4.20-4.36 (m, 4H), 4.70 (t, J = 2.2 Hz, 1H), 7.06-7.27 (m, 5H). 13C NMR
(62.5 MHz, CDCl3) d 19.9,
25.5, 26.6, 26.7, 41.0, 61.1, 64.2, 74.9, 77.5, 79.1, 113.6, 127.0, 128.3 (2C), 130.1
(2C), 136.2, 171.3, 173.1, 204.9. Anal. Calcd for C20H24O7:
C 63.82, H 6.43. Found: C 63.70, H 6.48. 9a: white solid; mp 67-68oC.; [
a ]D = -39 (c =
1.03, CHCl3); IR (KBr) 1792 (s), 1750 (s), 1715 (s) cm-1; 1H
NMR (250 MHz, CDCl3) d 1.37 ( s , 3H), 1.38 ( s, 3H), 1.44 (s, 3H), 2.10 (s, 3H), 2.94 (d, J = 13.1 Hz, 1H),
3.31 (d, J = 13.1 Hz, 1H), 4.07 (dd, J = 12.4 and 2.7 Hz, 1H), 4.16 (d, J
= 5.8 Hz, 1H), 4.36 (dd, J = 12.4 and 2.7 Hz, 1H), 4.44 (d, J = 5.8 Hz, 1H),
4.67 (t, J = 2.7 Hz, 1H), 7.12-7.22 (m, 5H); 13C NMR (62.5 MHz, CDCl3)
d 18.5, 25.4, 26.3, 26.5, 40.8,
61.0, 64.2, 74.9, 77.5, 79.2, 113.7, 127.1, 128.4 (2C), 130.0 (2C), 135.8, 171.3, 173.1,
204.7; Anal. Calcd for C20H24O7: C 63.82, H 6.43. Found:
C 63.86, H 6.55.
[19] 8a: mp 101-103oC.;
9a: mp 67-68oC.; 8b: mp 94-95oC.; 9b: mp 95-96oC.;
8d: mp 108-109oC.; 9d: mp 105-107oC.; 8e: mp
64-65oC.; 9e: mp 72-74oC.; 8f: mp 38-40oC.;
9f: mp 112-114oC.
[20] General
Procedure for Transesterification Reaction. A solution of 8a (1.00 g, 2.6 mmol)
and Ti(OEt)4 (1.21 g, 5.3 mmol) in ethanol (40 mL) was refluxed for five hours.
The solution was evaporated and the crude solid was purified by column chromatography on
silica gel under pressure, eluting with a mixture of hexanes: ethyl acetate. Product 13a
was obtained as a colorless oil (0.57 g , 92%): bp 125oC. (0.07 mmHg); [ a ]D = 66 (c = 0.64, CHCl3)
( [ a ]D= 62.5 (c =
0.42, CHCl3)11,21); IR (film) 2994, 1743 (s), 1715 (s) cm-1;
1H NMR (250 MHz, CDCl3) d 1.25 (t, J = 6.6 Hz, 3H), 1.29 (s, 3H), 2.17 (s, 3H), 3.05 (d, J
= 13.9, 1Hz), 3.27 (d, J = 13.9 Hz, 1H), 4.18 (m, 2H), 7.06-7.27 (m, 5H); 13C
NMR (62.5 Hz, CDCl3) d 13.9, 18.9, 26.4, 40.3, 60.7, 61.3, 126.8, 128.2 (2C), 130.1 (2C),
136.4, 172.3, 205.3; Anal. Calcd for C14H18O3: C 71.77, H
7.74. Found: C 71.89, H 7.95.
[21] Kato, K.; Suemune,
H.; Sakai, K Tetrahedron 1994, 50, 3315-3326.
[22] General
Procedure for Schmidt Rearrangement. Methanesulfonic acid (5.8 mL) was dropwise added
to a stirred mixture of ketoester 13d (1.30 g, 4.6 mmol) and DME (5 mL) cooled at
–30oC. Sodium azide (0.89 g, 13.7 mmol) was then added portionwise.
When the evolution of nitrogen ceased the mixture was left at room temperature 24h. More
DME (10 mL) and 30% aqueous ammonia were added till pH ca. 9. The mixture was partitioned
between dichlorometane and water. The organic layer was dried and evaporated. The residue
was purified through silica-gel eluting with hexanes: ethyl acetate mixtures of increasing
polarity to afford 14d (1.13 g, 82%) as an oil: bp 200oC (0.07 mmHg); [ a ]D = 51(c = 0.97, CHCl3);
IR (film) 3290 , 1736, 1652 cm-1; 1H NMR (250 MHz, CDCl3)
d 1.24 (t, J = 7.3 Hz,
3H), 1.60 (s, 3H), 1.86 (s, 3H), 3.28 (d, J = 13.8 Hz, 1H), 3.56 (d, J =
13.8 Hz, 1H), 4.15 (q, J = 7.3 Hz, 2H), 6.10 (broad s, 1H), 7.07-7.11(m, 1H),
7.33-7.39 (m, 2H), 7.43 (s, 1H), 7.62-7.72 (m, 3H); 13C NMR (62.5 Hz, CDCl3)
d 14.1, 23.4, 23.9, 40.9, 61.1,
61.8, 125.6, 125.9, 127.5 (2C), 128.0, 128.6, 132.3, 133.3, 134.1, 169.6, 173.9; Anal.
Calcd for C18H21NO3: C 72.22, H 7.07, N 4.68. Found: C
71.81, H 7.34, N 4.33.
[23] Gálvez, N.;
Moreno-Mañas, M.; Sebastián, R. M.; Vallribera, A. Tetrahedron 1996, 52,
1609-1616.
[24] 14a: bp 175oC./0.07
mmHg; 14b: mp 69-71oC.; 14d: bp 200oC./0.07 mmHg; 14e:
mp 88-90oC.; 14f: mp 72-74oC.
[25] 15a: white
solid; mp 160-164oC. (d); Anal. Calcd for C10H14NO2Cl:
C 55.69, H 6.54, N 6.49. Found: C 55.52, H 6.62, N 6.07. 15b: white solid; 161-163oC.
(d); Anal. Calcd for C10H13BrNO2Cl: C 40.77, H 4.45, N
4.75. Found: C 40.60, H 4.38, N 4.52. 15d: white solid; mp 170-174oC.
(d); Anal. Calcd for C14H16NO2Cl.H2O.: C
59.26, H 6.39, N 4.94. Found: C 59.12, H 6.41, N 4.83. 15e: white solid; mp 169-173oC
(d); Anal. Calcd for C13H19BrClNO2.1/2H2O: C
45.16, H 5.79, N 4.05. Found: C 44.88, H 5.80, N 3.94; 15f: white solid; 110-112oC.
(d); Anal. Calcd for C17H22NO2Cl.H2O:
C 62.66, N 4.30. Found: C 62.88, N 4.59.
[26] Moreno-Mañas, M.;
Sebastián, R. M.; Vallribera, A. Molins, E.; Espinosa, E. Tetrahedron: Asymmetry 1997,
8, 1525-1527.
[27] Mp's (oC):
17b: 52-54; 17d: 131-133; 17e: 133-135; 17f: 87-88.
All comments on this poster should be sent by e-mail to (mailto:[email protected]
ona.edu)
[email protected]
with A0006 as the message subject of your e-mail.