[A0014]
![]()
![]()
Received: 15 August 1999 / Uploaded: 26 August 1999
![]()
![]() Compounds containing
phosphine and phosphinimide units provide bifunctionality from a "soft"
phosphorus(III) center (which is predisposed for combination with soft metals) and a
"hard" nitrogen center (which is predisposed for combination with early
transition metals) so that the complex formation process is favored in a variety of
situations.
Compounds containing
phosphine and phosphinimide units provide bifunctionality from a "soft"
phosphorus(III) center (which is predisposed for combination with soft metals) and a
"hard" nitrogen center (which is predisposed for combination with early
transition metals) so that the complex formation process is favored in a variety of
situations.
![]() Ten years ago it was reported[1] that tungsten and molybdenum carbonyls react with N-trimethylsilyl[(diphenylphosphino)methyl]
diphenylphosphinimide 1 to give the cyclometallophosphinimide phosphanes 2
and 3.
Ten years ago it was reported[1] that tungsten and molybdenum carbonyls react with N-trimethylsilyl[(diphenylphosphino)methyl]
diphenylphosphinimide 1 to give the cyclometallophosphinimide phosphanes 2
and 3.

![]() In the course of our current
research on the chemistry of [(diphenylphosphino)methyl]diphenyl phosphinimides we
attempted a similar coordination of some N-aryl derivatives 4 under similar
reaction conditions. In our hands, such small variation of the starting phosphinimides (N-aryl
instead of N-trimethylsilyl) with respect to the scheme above resulted in notably
different reaction products. Thus, the reaction of N-aryl[(diphenylphosphino)methyl]diphenylphosphinimides
4[2] with W(CO)5THF[3]
gave rise to the previously unknown tungsten complexes 5 albeit in low yields.
Compounds 5 precipitated out from the reaction medium as yellow solids and were
isolated by filtration in virtually pure state.
In the course of our current
research on the chemistry of [(diphenylphosphino)methyl]diphenyl phosphinimides we
attempted a similar coordination of some N-aryl derivatives 4 under similar
reaction conditions. In our hands, such small variation of the starting phosphinimides (N-aryl
instead of N-trimethylsilyl) with respect to the scheme above resulted in notably
different reaction products. Thus, the reaction of N-aryl[(diphenylphosphino)methyl]diphenylphosphinimides
4[2] with W(CO)5THF[3]
gave rise to the previously unknown tungsten complexes 5 albeit in low yields.
Compounds 5 precipitated out from the reaction medium as yellow solids and were
isolated by filtration in virtually pure state.

![]() As shown in the scheme the
coordination of the metal to the P(III) center was successfully accomplished but the
phosphinimide function did not survive but hydrolyzed to the phosphine oxide and the
corresponding aniline. At the present we can not say unequivocally if the nitrogen atom of
the phosphinimide unit become or not coordinated to the metal previously to the hydrolytic
process, but it seems quite clear that such coordination would enhance the susceptibility
of the phosphinimide to the hydrolysis by making the P(V) atom more electrophilic. In the
absence of the tungsten carbonyl W(CO)5THF compounds 4 did not show such
hydrolytic sensitivity[4].
As shown in the scheme the
coordination of the metal to the P(III) center was successfully accomplished but the
phosphinimide function did not survive but hydrolyzed to the phosphine oxide and the
corresponding aniline. At the present we can not say unequivocally if the nitrogen atom of
the phosphinimide unit become or not coordinated to the metal previously to the hydrolytic
process, but it seems quite clear that such coordination would enhance the susceptibility
of the phosphinimide to the hydrolysis by making the P(V) atom more electrophilic. In the
absence of the tungsten carbonyl W(CO)5THF compounds 4 did not show such
hydrolytic sensitivity[4].
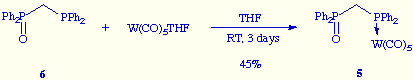
![]() We have established the
structure of 5 by its analytical and spectroscopic data as well, as presumed,
through its alternative preparation by reaction of
[(diphenylphosphino)methyl]diphenylphosphine oxide 6 with W(CO)5THF.
We have established the
structure of 5 by its analytical and spectroscopic data as well, as presumed,
through its alternative preparation by reaction of
[(diphenylphosphino)methyl]diphenylphosphine oxide 6 with W(CO)5THF.
![]()
![]() The infrared spectrum shows
absorption bands corresponding to overlapping E and A1(1) modes at
1935 cm-1, A1(2) mode at 2074 cm-1 and B1
mode at 1983 cm-1.
The infrared spectrum shows
absorption bands corresponding to overlapping E and A1(1) modes at
1935 cm-1, A1(2) mode at 2074 cm-1 and B1
mode at 1983 cm-1.
![]() The methylene hydrogen atoms
appear in the 1H NMR as a double doublet at d 3.66
ppm with 2JH-P coupling constants of 7.8 Hz and 11.6 Hz. By
contrast, compounds 4 and 6 do not show coupling of their methylene protons
with the P(III) atom, but only with the P(V) one (4: d
3.22-3.24 ppm, 2JH-P = 12.6-12.9 Hz; 6: d 3.09 ppm, 2JH-P = 12.9 Hz).
The methylene hydrogen atoms
appear in the 1H NMR as a double doublet at d 3.66
ppm with 2JH-P coupling constants of 7.8 Hz and 11.6 Hz. By
contrast, compounds 4 and 6 do not show coupling of their methylene protons
with the P(III) atom, but only with the P(V) one (4: d
3.22-3.24 ppm, 2JH-P = 12.6-12.9 Hz; 6: d 3.09 ppm, 2JH-P = 12.9 Hz).
![]() The CO region of the 13C
NMR spectrum consists of a set of downfield peaks assigned to the CO group trans to
the coordinated phosphorus atom (d 200.4 ppm, 2JC-P
= 22.2 Hz) and a set of upfield signals arising from the cis CO groups (d 198.0 ppm, 2JC-P = 6.8 Hz and 4JC-P
= 2.9 Hz).
The CO region of the 13C
NMR spectrum consists of a set of downfield peaks assigned to the CO group trans to
the coordinated phosphorus atom (d 200.4 ppm, 2JC-P
= 22.2 Hz) and a set of upfield signals arising from the cis CO groups (d 198.0 ppm, 2JC-P = 6.8 Hz and 4JC-P
= 2.9 Hz).
![]() The 31P NMR
spectrum shows two set of signals: one at d 24.17 ppm a doublet
for the P(V) atom with a coupling constant of 2JP-P' = 3.7
Hz, and another set centered at d 3.87 ppm due to the
phosphorus atom coordinated to the tungsten atom with the following coupling constants 2JP-P'
= 3.7 Hz, 1JP-W = 248.6 Hz.
The 31P NMR
spectrum shows two set of signals: one at d 24.17 ppm a doublet
for the P(V) atom with a coupling constant of 2JP-P' = 3.7
Hz, and another set centered at d 3.87 ppm due to the
phosphorus atom coordinated to the tungsten atom with the following coupling constants 2JP-P'
= 3.7 Hz, 1JP-W = 248.6 Hz.
![]() As a continuation of this
work we are now trying to achieve the formation of the new metallacycle 7 by
intramolecular displacement of a carbonyl ligand on the tungsten by the phosphine oxide
function:
As a continuation of this
work we are now trying to achieve the formation of the new metallacycle 7 by
intramolecular displacement of a carbonyl ligand on the tungsten by the phosphine oxide
function:
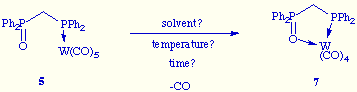
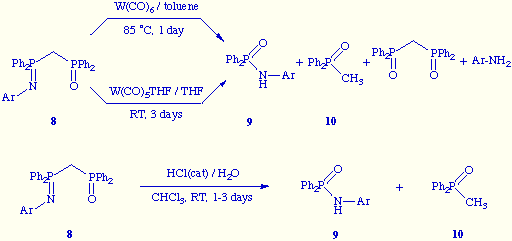
![]() We also have attempted to
prepare other new tungsten complexes by using [(diphenylphosphinoyl)methyl]diphenyl
phosphinimides 8 but without success. Thus, the reaction of 8 with W(CO)6
or W(CO)5THF did not give a tungsten complex by coordination of the metal to
the oxygen or nitrogen atoms or both; instead of, these reactions gave rise mostly to the
fragmentation of the putative ligands, yielding mainly phosphoranilides 9 and
diphenylmethylphosphine oxide 10, along with some minor amounts of the products
resulting from the hydrolysis of the phosphoramide unit of 8. The fragmentation of
compounds 8 to give 9 and 10 has been previously observed in our lab
when those compounds were treated with catalytic amounts of HCl in CHCl3
solution, and an explanation for such P-C cleavage has been acquainted[4]. Taking this precedent into account, it seems that W(CO)6
and W(CO)5THF play the role of simple Lewis acid catalysts in the here shown
fragmentation processes.
We also have attempted to
prepare other new tungsten complexes by using [(diphenylphosphinoyl)methyl]diphenyl
phosphinimides 8 but without success. Thus, the reaction of 8 with W(CO)6
or W(CO)5THF did not give a tungsten complex by coordination of the metal to
the oxygen or nitrogen atoms or both; instead of, these reactions gave rise mostly to the
fragmentation of the putative ligands, yielding mainly phosphoranilides 9 and
diphenylmethylphosphine oxide 10, along with some minor amounts of the products
resulting from the hydrolysis of the phosphoramide unit of 8. The fragmentation of
compounds 8 to give 9 and 10 has been previously observed in our lab
when those compounds were treated with catalytic amounts of HCl in CHCl3
solution, and an explanation for such P-C cleavage has been acquainted[4]. Taking this precedent into account, it seems that W(CO)6
and W(CO)5THF play the role of simple Lewis acid catalysts in the here shown
fragmentation processes.
![]()
Thanks are given to Direccion General de Investigacion Cientifica y Tecnica (project number PB95-1019) and Acedesa (a division of Takasago) for financial support.
![]()
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0014 as the message subject of your e-mail.