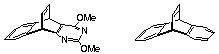[A0015]
|
|
New routes to dibenzobarrelene and pyrimidinobenzobarrelenes: a synthetic and computational study
Mirta Golic Douglas N. Butler Ronald N. Warrener* and Davor Margetic
*Professor Ron Warrener.
Director, Centre for Molecular Architecture, Central Queensland University,
Bruce Highway, ROCKHAMPTON QLD 4702
Australia
Fax: 61 7 49 309917, Tel.: 61 7 4930 9845, E-mail: [email protected]
Received: 3 August 1999 / Uploaded: 11 August 1999
Flash vacuum pyrolysis (FVP) and oxidative decarboxylation methods were used as a novel synthetic routes to dibenzobarrelene and pyrimidinobenzobarrelenes. A computational study of these reactions was conducted in order to gain more insight into the reaction mechanism. |
|
| 4. Experimental Details | |
| 5. References | |
1. Flash vacuum pyrolysis (FVP) routes to dibenzobarrelenes
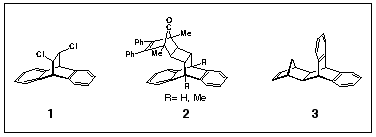
There are several methods reported for the synthesis of dibenzobarrelene 5, yet there is no convenient route to its production on a large scale. The best yield reported to date involves the dechlorination of adduct 1 formed by addition of 1,2-dichloethene to anthracene.1a While this route appears simple and dechlorination yields are high (sodium in n-amyl alcohol1b, 88 % yield or zinc1a 98 % yield), formation of adduct 1 in our hands is fickle and never clean (sealed tube, >200 oC). Another method involves the photochemical decomposition of hemicyclone adduct 22, however, preparation of the starting materials for use as photosubstrates is not trivial. Some years ago we reported the synthesis of adduct 3 by the addition of norbornadiene to anthracene.3 We noted that heating 3 above 400 o C produced anthracene 7 and dibenzobarrelene 5, but we did not follow up the reaction other than to note that the products were formed in roughly equal amounts. This result left open the question about the origin of anthracene as it could have arisen by the fragmentation of dibenzobarrelene by retro-Diels-Alder elimination of acetylene or it might have been formed directly from adduct 3 in a reaction corresponding to the reversal of its formation from 7 and 8.
Scheme 1
We have now returned to the thermolysis of 3 on three counts:
a) to investigate its potential as a short route to dibenzobarrelene
b) to study the competing retro-Diels-Alder processes under gas phase conditions4
c) to apply computational methods (semiempirical and ab initio) to evaluate the energetics
of the competing pathways involved in the fragmention of 3 and related adducts
The study has been expanded to include fragmentation of the isomeric anthacene/norbornadiene adduct 43 as it provides information on the stereoselectivity of the fragmentation pathway leading to dibenzobarrelene. As well, we have studied diaza-analogues of this FVP process as a route to a new class of pyrimidino-benzobarrelenes. Finally, we have investigated some alternative routes to these dibenzobarrelene and diaza-dibenzobarrelenes using decarboxylation and oxidative bisdecarboxylation methods.
Scheme 2
1.1 Dibenzobarrelene
Flash vacuum pyrolyses were conducted under high vacuum (0.01 - 0.001 mbar) in an unpacked 600 x 6 mm pyrex tube heated by a horizontally-mounted 'Thermolyne' model 21100 tube furnace. The optimum pyrolysis temperature (calibrated using a thermocouple attached externally to the glass pyrolysis tube) was determined separately for each substrate.
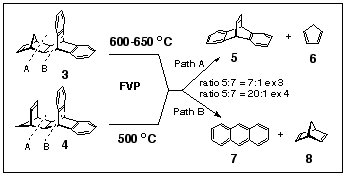
Our investigation has shown that adduct 4 undergoes decomposition under FVP conditions at lower temperatures (500 o C), while temperatures of 600 - 650 o C were required to crack adduct 3 (Scheme 2).
FVP of adduct 3 at 500 oC resulted in starting material being recovered, having sublimed through the pyrolysis tube unchanged. Raising the tube temperature to 600 o C (0.01 mbar) caused 85% breakdown of starting material to yield a 12:1 mixture of dibenzobarrelene 5 and anthracene 7. Complete cracking did not occur until 650 o C (0.01 mbar) and resulted in the production of a 7:1 mixture of 5:7. It is significant that the actual yield of dibenzobarrelene 5 is not significantly improved at the higher temperature as the proportion of anthracene increases as the temperature is raised.
FVP of isomer 4 was significantly complete (20% of 4 sublimed unchanged) at 500 o C (0.001 mbar) and produced a mixture of dibenzobarrelene 5 and anthracene 7 in a ratio of 20:1.
Separate FVP of dibenzobarrelene 5 at 700 o C (0.01 mbar) produced a mixture of 5 and 7 (1.7:1 ratio), thus demonstrating the significantly greater thermal stability of dibenzobarrelene 5 relative to either of it precursor adducts 3 or 4. This means that the production of anthracene comes mainly from the competing fragmentation pathway open to 3 or 4, rather than by secondary decomposition of 5.
1.2 Diazadibenzobarrelenes
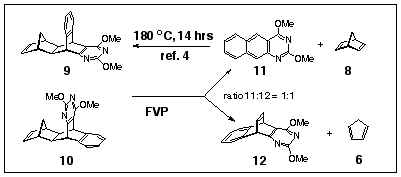
We have recently reported that the addition of norbornadiene 8 to 2,4-dimethoxy-1,3-diazaanthracene 11 yields a pair of [4+2] cycloadducts5 9 and 10 by addition at the central ring of 11. They differ only in the positioning of the benzene and pyrimidine rings in relation to the carbocyclic frame (Scheme 2). Each can serve as a precursor for the production of the same pyrimidinobenzobarrelene provided the loss of cyclopentadiene is energetically favourable. Accordingly, we have subjected the mixture of adducts 9 and 10 formed from the reaction of norbornadiene with 11 to FVP under similar reaction conditions as those used above (550 o C, 0.001 mbar). This experiment yielded a mixture of the diazaanthracene 11 and the pyrimidino barrelene 12 in approximately 1:1 ratio, when the pyrolysis tube was packed with cracked glass. A second experiment gave a 5:4 mixture of 11 and 12. Diazadibenzobarrelene 12 was isolated in 13% yield following selective solubility in methanol followed by radial chromatographic separation on silica.
Scheme 3
Proton NMR spectral data for 12 fully support the barrelene structure with two resonances for the bridgehead protons H10 and H9 (doublets of doublets at d 5.14 and 5.33, J=4.7, 2.7 Hz), similar to the chemical shifts for the analogous protons in the dibenzobarrelene 51 and adduct 13 prepared by the addition of dimethyl acetylene dicarboxylate to diazaanthracene 9 (Fig. 1). Singlet resonances at d 3.95 and d 3.97 were also observed for the methoxy groups in 12.
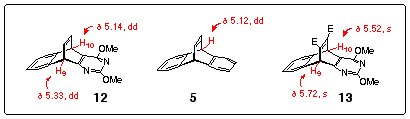
Figure 1
The signals for the alkene protons fall in the low field area (d 6.97-7.36 ppm) of the 1H NMR spectrum and overlap with the resonances for the aromatic protons. The 13C NMR spectrum of 12 contains four signals at d 42.7, 53.3, 53.5 and 54.6 which belong to the two saturated carbons C9, C10 and the two methoxy carbons. The remaining twelve signals positioned between 118.0 and 179.2 ppm correspond to the chemical shifts of the pyrimidine, aromatic and double bond carbon atoms. Finally, the mass spectrum of barrelene 12 has the molecular ion (m/z 266) as the base peak.
1.3 The Preparation of a Uracil-Containing Diazadibenzobarrelene
The corresponding uracil analogues 14 and 15 were prepared by hydrolysis of the afore-mentioned adducts 9 and 10.5 When the isomeric mixture of 14 and 15 was subjected to FVP (500 o C, 0.001 mbar) two products were formed: 1,3-dihydro-1,3-diazaanthracene-2,4-dione 17 as the major product and diazadibenzobarrelene 16 as the minor product in a 3:2 ratio (Scheme 4). Two doublets at d 4.87 (J= 5.7 Hz) and 5.23 ppm (J= 5.7 Hz) were assigned to the bridgehead protons H10 and H9 of uracilobenzobarrelene 16. The vinyl proton signals in all dibenzobarrelenes studied herein have overlapped with the aromatic protons thereby preventing their use as an indicator of barrelene formation using 1H NMR as the reaction monitor.
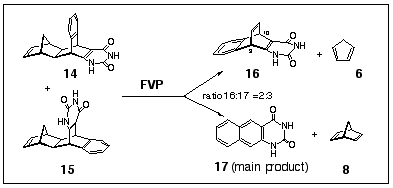
Scheme 4
2. Chemical Routes to Dibenzobarrelenes
2.1 The Oxidative Decarboxylation Method
Oxidative decarboxylation is a recognised method for producing unsaturated ring-structures from vicinal dicarboxylic acid precursors.6 Accordingly, we have prepared the maleic anhydride adducts of diazanthracene 11 by heating the reagents together in refluxing toluene. Treating the resultant mixture of exo-adduct 19 (exo-descriptor related to the benzene ring) and endo-adduct 20 with Ni(CO)2(PPh3)27 (200 o C, diglyme, 30 minutes) formed the diazadibenzobarrelene 2 in 38% yield (Scheme 5). A significant by product in this reaction is the diazaanthracene 11, which is formed (14%) by competitive retro-Diels-Alder fragmentation of the anhydride adducts under the high temperatures required for the reaction.
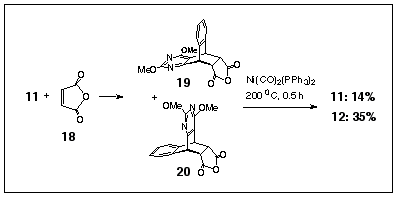
Scheme 5
Application of these same reaction conditions to the anthracene / maleic anhydride adduct 21 produced dibenzobarrelene 5 in 82% yield (Scheme 6). As the starting adduct 21 is so readily prepared in quantity, this method is the best route to dibenzobarrelene 5 yet reported. Wolinsky has used lead tetraacetate decarboxylation of diacid obtained by hydrolysis of 21 to prepare 5 in 18% yield,8 as well as decarboxylation of the anthracene / fumaric acid adduct with lead tetraacetate to produce 5 in 29% yield.8
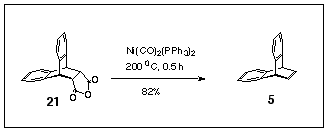
Scheme 6
2.2 The Decarboxylation Method
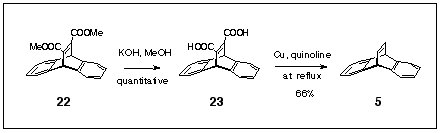
Scheme 7
Copper-catalysed decarboxylation is a recognised method for the conversion of carboxylic acids to hydrocarbons.6 Accordingly, we prepared the adduct 22 from anthracene and dimethyl acetylenedicarboxylate using the literature method and hydrolysed it to the barrellene diacid 23 in essentially quantitative yield using KOH in methanol (Scheme 7). Refluxing this diacid in quinoline containing a catalytic amount of copper powder gave dibenzobarrelene 5 in 66% isolated yield. This two-step procedure (a cycloaddition and a hydrolysis) starting from commercially available reagents (anthracene and dimethyl acetylenedicarboxylate) must represent the most cost effective way to produce dibenzobarrelene 5 yet reported. Figeys9 prepared the dibenzobarrelenes bearing substituents sensitive to other eliminative procedures in high yields using the same prodecure. The method has not yet been applied to the diazadibenzobarrelenes, but must be considered suitable for the formation of the uracil-containing barrelene 16. By using the DMAD adduct 13 as the starting material, hydrolysis should convert the pyrimidine to the uracil at the same time as converting the ester groups to acids.
3. Computational Study of the Pyrolysis Pathway
This section of the paper deals with the energetics of the competing pathways (pathway A and pathway B) for fragmentation of norbornadiene adducts of type 3 and 4. Path A is a retro-Diels-Alder reaction involving the loss of cyclopentadiene to yield dibenzobarrelene 5; Path B is the retro-Diels-Alder cleavage of 3 and 4 to afford norbornadiene 8 and anthracene 7 (cf, Scheme 2). Similar studies have been conducted on the pyrimidine-containing adducts 9,10 and 14,15. The retro-Diels-Alder reaction of dibenzobarrelene 5 to form anthracene and acetylene has also been investigated, as it provides an alternative source of anthracene in these thermally-induced fragmentation reactions.
We have estimated the activation energies for all transition states (TS) for all fragmentations using semiempirical (AM1 and PM3), ab initio (RHF/3-21G, RHF/6-31G*//3-21G), as well as single point energy estimation by the DFT method on the ab initio optimised structures of TS's (B3LYP/6-31G*//3-21G).
All the calculation methods employed predict that a smaller activation energy is required for the cyclopentadiene elimination from 3 or 4 than for norbornadiene elimination (Scheme 8), which is in full accordance with experiment. Furthermore, smaller thermal stability of 4 relative to 3 is also correctly predicted at all theoretical levels. Dibenzobarrelene 5 can be further converted into anthracene 7 by acetylene elimination via TS6, but the activation energy is higher than TS1 - TS4. These computational results suggest that anthracene is formed mainly through TS2 since it requires a lower activation energy. Norbornadiene can further fragment to acetylene and cyclopentadiene via the TS5.
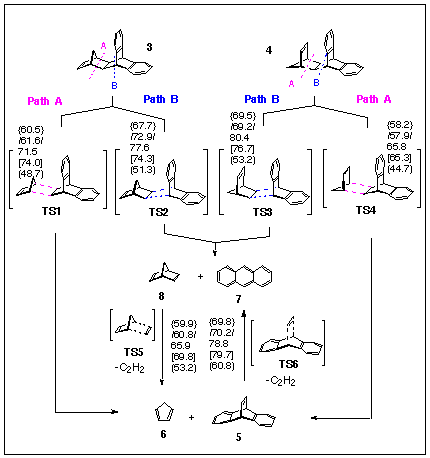
Scheme 8. {AM1}, /PM3/, RHF/3-21G, [RHF/6-31G*//3-21G] and (B3LYP/6-31G*//3-21G) activation energies, in kcal/mol.
Due to difficulties with conformational flexibility of the studied molecules (methoxy rotations), the computational study of fragmentation of 9, 10 was conducted only at the semiempirical level (Scheme 8). In this reaction, AM1 transition state energies wrongly predict that the cyclopentadiene elimination is preferred over norbornadiene elimination by 6-7 kcal/mol.
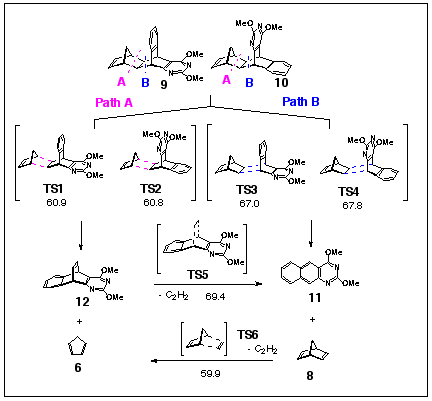
Scheme 9. AM1 activation energies, in kcal/mol.
Finally, a computational study of FVP of uracils 14 or 15 predicts (at all computational levels employed) that norbornadiene elimination has a smaller activation barrier, leading to formation of naphthouracil 17 as the dominant product (Scheme 10). This prediction is again in full accordance with experiment, where 17 is major product. The only exception is the AM1 method predicting very similar activation barriers for cyclopentadiene and norbornadiene eliminations (difference is 0.4 - 0.6 kcal/mol).
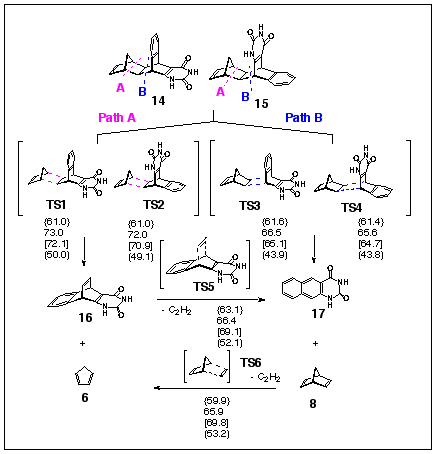
Scheme 10. {AM1}, /PM3/, RHF/3-21G, [RHF/6-31G*//3-21G] and (B3LYP/6-31G*//3-21G) activation energies, in kcal/mol.
Acknowledgements. M. G. thanks the Centre for Molecular Architecture for the award of a PhD scholarship.
Flash-vacuum pyrolysis experiments were conducted under vacuo (0.001 mbar) in an 600x6 mm pyrex tube heated by a horizontally-mounted 'Thermolyne' model 21100 tube furnace using 100 - 200 mg of substrate. In some experiments the pyrex tube was packed with broken soda-glass pieces. Products were collected at the end of furnace on cooler part of the tube. Volatile products (cyclopentadiene and norbornadiene were condensed in liquid nitrogen trap). Pyrolyses over 600 o C were conducted under lower vacuum (0.01 mbar) due to deformations of the pyrex glass tubes at higher vacuum. No experiments were conducted using silica thermolysis tubes.
9,10-Etheno-9,10-dihydroanthracene (Dibenzobarrelene) 5
Method A FVP of 4 (120 mg) at 500 o C gave mixture of 4:5:7 in 1:4:0.2 ratio (93 mg). When the same compound was heated in an open tube (at 500 o C, several minutes) at atmospheric pressure a mixture of 4:5:7 in 1:5.4:0.7 ratio was obtained.
FVP of 3 (150 mg) at 600 o C (higher pressure 0.01 mbar) produced a mixture of 1:5:7 in 1:6.1:0.5 (115 mg). FVP of 3 (110 mg) at 650 o C (higher pressure 0.01 mbar) produced a mixture of 3:5:7 in 0:1:0.14 ratio (85 mg).
FVP of 5 at 700 o C (higher pressure 0.01 mbar) produced a mixture of dibenzobarrelene 5 and diazaanthracene 7 (1.7:1).
Method B. Dibenzobarrelene 5 was obtained by oxidative decarboxylation of the anthracene - maleic anhydride adducts (500 mg, 1.81 mmol) by reaction with Ni(CO)2(PPh3)2 (1.4 g, 2.25 mmol) in anhydrous diglyme (5 ml) at 165 o C for two hours. The catalyst was filtered off on a celite column and 3 (359 mg), together with small amount of the catalyst, washed off the column with petroleum ether. Final separation was obtained by radial chromatography using the petroleum ether/ethyl acetate 20:1 solvent mixture for elution yielding white powder (305 mg, 82%, m.p. 118-119 o C lit. 119-120.5 oC).
1H NMR (300 MHz, CDCl3) d 5.12 (narrow m, 2H), 6.94 (m, 4H), 7.00 (m, 2H).
(9a,10a)-9,10[1',2']Benzeno-9,10-dihydro-2,4-dimethoxyquinazoline, Diazadibenzobarrelene 12
Procedure A: A mixture of isomers 9 and 10 (3:2 ratio, 650 mg, 1.96 mmol) was placed in a pyrex-glass packed tube and flash vacuum pyrolised at 550 o C and 0.4 mbar. The products (416 mg of crude mixture) were trapped on an ice-water cooled section of the pyrolysis tube and partially separated by selective solubility in methanol, whereby 9 was precipitated. 2,4-Dimethoxy-1,3-diazadibenzobarrelene (12, m.p. 103-104 o C, 66 mg, 13% yield) was isolated by radial chromatography with petroleum ether/ethyl acetate employing gradient elution technique.
Procedure B: The title compound 12 was obtained in 38% yield from the oxidative decarboxylation of a mixture of anhydrides 19 and 20 (334 mg, 0.99 mmol) by reaction with Ni(CO)2(PPh3)2 catalyst (930 mg, 1.50 mmol) in anhydrous diglyme (4 ml) at 165 o C for 30 minutes. The catalyst was filtered off on a celite column and product 12 washed from the column with petroleum ether. Recrystallisation from dichloromethane/methanol yielded 12 (119 mg). Diazaanthracene 11 (34 mg 14%) was obtained as a byproduct.
1H NMR (300 MHz, CDCl3) d 3.95 (s, 3H), 3.97 (s, 3H), 5.14 (dd, J=4.7, 2.7 Hz, 1H), 5.33 (dd, J=4.7, 2.7 Hz, 1H), 7.00 (m, 4H), 7.33 (m, 2H). 13C NMR (75 MHz, CDCl3) d 42.7, 53.3, 53.5, 54.6, 118.0, 122.9, 124.1, 124.6, 125.0, 138.3, 139.9, 144.5, 145.6, 162.2, 163.4, 179.2. MS, m/z (%) 266 (M+, 100), 251 (M+-CH3, 25), 240 (2,4-dimethoxy-1,3-diaza-anthracene, 15), 193 (M+-NHCOCH3, 41), 166 (193-CHCH2, 23). HRMS (EI) for C16H14N2O2 requires for M+ (m/z) 266.1055, found: 266.1056
(9a,10a)-9,10[1',2']Benzeno-1,3,9,10-tetrahydro-quinazolin-2,4-dione, Diazadibenzobarrelene 16.
Procedure A: FVP of an isomeric mixture of 14 and 15 (3:2 ratio, 20 mg, 0.06 mmol) at 500 o C yielded 1,3-dihydro-1,3-diazaanthracene-2,4-dione (naphthouracil) 17 as the major product and 2,4-dioxo-1,3-diazadibenzobarrelene 16 as the minor isomer (3:2 ratio).
Procedure B: Dimethoxydiazadibenzobarrelene 12 (15 mg, 0.057 mmol) was treated with hot hydrochloric acid (2 ml of 5M HCl, at 100 o C) for two hours. Evaporation of acid under reduced pressure yielded a white solid, which was washed with dichloromethane to remove unreacted 12, to afford the title barrelene 16 (9 mg) in 66% yield, m.p. 183-186 o C.
1H NMR (300 MHz, 20% DMSO-d6 in CDCl3) d 5.01 (d, J=5.6 Hz), 5.25 (d, J=5.6 Hz), 6.88 (narrow m, 2H), 6.96 (m, 2H), 7.07 (m, 2H). 1H NMR (400 MHz, 20% CD3OD in CDCl3) d 4.87 (d, J=5.7 Hz), 5.23 (d, J=5.7 Hz), 6.82 (m, 1H), 6.91 (m, 2H), 7.03 (m, 1H), 7.22 (m, 2H,). 13C NMR (100 MHz, 20% CD3OD in CDCl3) d 42.5, 49.2, 116.5, 122.7, 123.2, 124.1, 125.1, 136.0, 142.1 (overlap), 142.7, 146.3, 161.0, 163.7. HRMS (EI) for C16H14N2O2 requires for M+ (m/z) 238.0742, found (m/z) 238.0741. (Signals for the NH protons were not observed in the 1H NMR spectra, as the exchange between these protons and NMR deuteriated protic solvents had occured).
Computational details:
Semiemprical and ab initio calculations were performed using the SPARTAN suite of programs10 on a Silicon Graphics R10000 indigo workstation. DFT11 single point energies on 3-21G optimised structures were estimated using the GAUSSIAN98 program12 operating on a Silicon Graphics Oxygen R5000 workstation.
[1] a) Cristol, S. J.; Lim, W. Y. J. Org. Chem. 1969, 34, 1; b) Cristol, S. J.; Bly, R. K. J. Am. Chem. Soc. 1960, 82, 6155; c) Cristol, S. J.; Hause, N. L. J. Am. Chem. Soc. 1952, 74, 2193.
[2] Nunn, E. E.; Warrener, R. N. Synth. Commun. 1972, 2, 61.
[3] Butler, D. N.; Barrette, A.; Snow, R. A. Synth. Commun. 1975, 5, 101.
[4] Wiersum, U. E. Recl. Trav. Chim. Pays-Bas. 1982, 101, 317, 365; Wiersum, U. E. Aldrichimica Acta, 1981, 14(3), 53; Brown, R. F. C. Pyrolytic methods in Organic Chemistry, Vol 41, Organic Chemistry Monographs, Academic Press, New York, 1980.
[5] Warrener, R. N.; Golic, M.; Butler, D. N. Synlett 1997, 1105.
[6] De Lucchi, O.; Modena, G. Tetrahedron 1984, 40, 2585.
[7] Trost, B. M.; Chen, F. Tetrahedron Lett. 1971, 2603; Dauben, W. G.; Rivers, G. T.; Twieg, R. J.; Zimmerman, W. T. J. Org. Chem. 1976, 41, 887.
[8] Cimarusty, C. M.; Wolinsky, J. J. Am. Chem. Soc. 1968, 90, 113.
[9] Figeys, H. P.; Dralants, A. Tetrahedron 1972, 28, 3031.
[10] SPARTAN Version 5.0, Wavefunction Inc. 18401 Von Karman Ave. Suite 370, Irvine, California, 1997.
[11] Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989; Ziegler, T. Chem. Rev. 1991, 91, 651.
[12] Gaussian 98, Revision A.5, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1998.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0015 as the message subject of your e-mail.