 |
|
[A0016]
|
|
Functionalised Rigid Rods - Dream or Reality
Davor Margetic Martin R. Johnston Ronald N. Warrener*
*Professor Ron Warrener.
Director, Centre for Molecular Architecture, Central Queensland University,
Bruce Highway, ROCKHAMPTON QLD 4702
Australia
Fax: 61 7 49 309917, Tel.: 61 7 4930 9845, E-mail: [email protected]
Received: 3 August 1999 / Uploaded: 11 August 1999
|
|
|
|
|
|
|
|
|
During the past several years molracs1 have been employed as the structural backbone for the positioning of various moieties in well defined geometries. These have included such entities as pyrimidines2, phenanthrolines3,4, crown ethers5 and porphyrins2. The synthesis of these backbones has evolved from serial cycloaddition strategies into those incorporating functionalised building BLOCKs and appropriate coupling reactions6,7. With the stereoselectivity of the oxadiazole (OD), ACE and s-tetrazine based coupling reactions well established, the design of larger supramolecular structures using the molrac concept now centres around building BLOCK design. In particular, the shape and positioning of functionality within the BLOCK is all important since these characteristics are retained in the target molrac.
A particularly relevant variation on the usual curved molrac topology was the inclusion of the pentacyclic bis-norbornene unit 1 to create rigid rods8. Application of the various coupling strategies mentioned above facilitated the synthesis of extended rods with little or no curvature.
As a further extension of this methodology, we sought a BLOCK that would allow functionality to be positioned in the all important 7- position of the norbornene skeleton as well as facilitate the synthesis of linear rigid systems utilising the known coupling procedures. Herein we report that 12 has been identified as a BLOCK fulfilling the above requirements and discuss its synthesis as well as several attempts to incorporate 12 into various linear architectures.
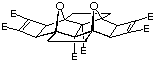
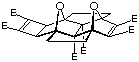
Molecular modelling is an invaluable aid in determining suitable materials for inclusion into molrac structures. Several BLOCK candidates are shown in Figure 1 and contain various degrees of curvature as indicated by the comparison between the calculated (AM1) C-C and H-H distances shown. The curvature of the system may be expressed in the difference between these distances, with a large difference implying high curvature. The Paquette-based system 18, containing two one carbon bridges directly linked, possesses the least curvature of any of the BLOCKs shown, whereas the bisnorbornane skeleton 2 has the highest. The inclusion of oxygen in BLOCK 3 still results in a certain degree of curvature. However, the Cram-based system 4,10 while still containing two functionalities, is still reasonably linear in geometry and represents an excellent combination of functionality and shape. The system 4 was thus identified as a suitable target for inclusion within the molrac structure.

Figure 1: Molecular models of the various BLOCKs carried out at the AM1 level of theory.
Distances are in Angstroms (Å).
The synthesis of 12 followed that first developed by Cram10 and is outlined in Schemes 1 and 2. Treatment of 2-methylfuran 5 under Mannich conditions resulted in the isolation of the adduct 6 which was subsequently quatternised with methyl iodide to yield 7, which after being subject to counterion exchange produced 8 (Scheme 1). Pyrolysis of 8 (via Hoffman elimination) resulted in the formation of the transient exocyclic diene 9, which dimerises in situ to yield the product bisfuranocyclophane 10 in reasonable yield. The reaction sequence was carried out on a multi-gram scale.
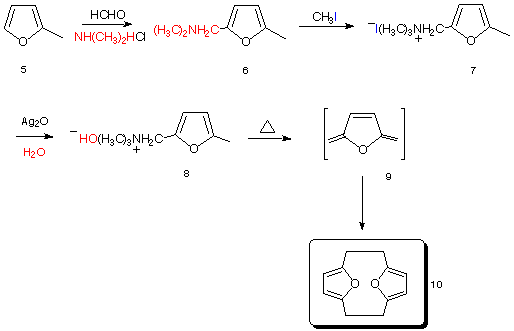
Scheme 1
Once isolated, the furanocyclophane 10 was allowed to react with dimethylacetylene dicarboxylate 11 (DMAD) to give the pincer cycloaddition adduct 12 (Scheme 2). The intermediate 1:1 adduct between 11 and DMAD has the possibility of forming both "pincer" and "domino" adducts, yet the "pincer" adduct 12 was the only isomer observed.
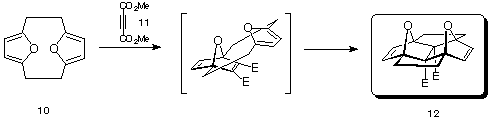
Scheme 2
The curvatures of the various molrac structures were initially examined by molecular modelling at the AM1 level of theory. Shown in figures 2, 3 and 4 are various modelled structures based on the 1, 2 and 4 BLOCKs. In all instances the pronounced curvature of the molecules constructed with the 2 BLOCK is apparent. In contrast the use of 1 or 4 creates a more linear system with 1 producing more linear structures than 4.
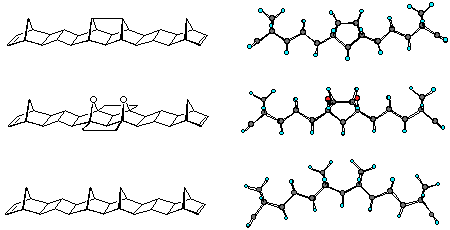
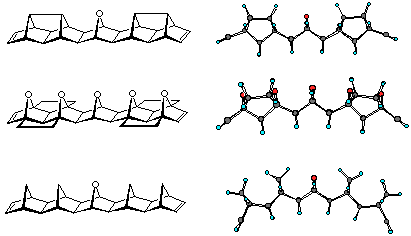
Figure 3: Oxadiazole or ACE coupled products

Figure 4: Tetrazine coupled adducts
The first coupling method to be examined using 12 was the epoxide based ACE procedure11. While the stereoselectivity of the ACE reaction is well known for norbornene terminated substrates, the use 7- oxa containing BLOCKs results in the isolation of both exo,exo- and exo,endo- adducts12. Thus, for the stereoselective use of 12 the epoxide functionality must be contained within 12 and this necessitated the synthesis of 14 via the DMAD adduct 13 (Scheme 3). However, several attempts to prepare 13 have proved ineffective with only very minor amounts of material produced. Instead a complex mixture containing 15, 16 and 17 along with other unidentified products. Such materials presumably result from an intermediate 18 (by analogy with the s-tetrazine coupling reaction intermediate, Scheme 4) to produce 15 which subsequently undergoes reaction with DMAD under the reaction conditions to form the tetraester 16. Further reaction of 16 with DMAD under ruthenium catalysed conditions yields the hexaester 17. The conversion of 16 to 17 has been verified in a separate experiment starting with pure 16.
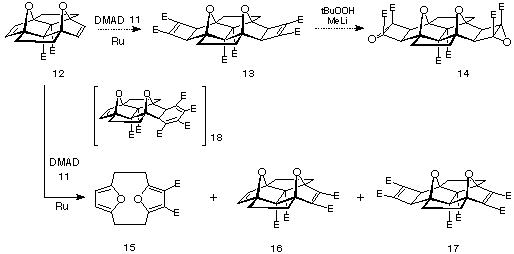
Scheme 3
Cram has indeed reported that 12 undergoes a partial retro Diels-Alder reaction at 100ûC resulting in the formation of an equilibrium between 12 and the bisfuranocyclophane 10 after a 3-4 hours. Further attempts at the synthesis of 13 utilising lower reaction temperatures were unsuccessful.
The thermal instability of 12 eliminated the possibility of the OD coupling procedure (140û, 20 hours, sealed tube) and hence attention was turned to the use of s-tetrazine as a coupling reagent since low temperatures and high pressures have been found to be favourable for its use13. The proposed synthesis of molrac 20 using tetrazine is outlined in Scheme 4.
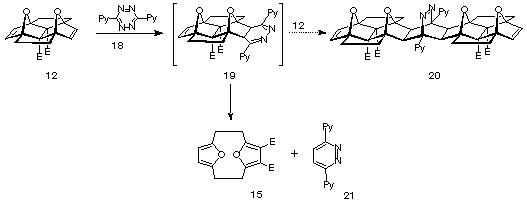
Scheme 4
Combination of two equivalents of 12 with one equivalent of s-tetrazine 18 in DCM revealed the loss of the characteristic red colour of 18 within several minutes, an phenomena usually expected to be associated with the rapid formation of the intermediate dihydropyrazine 19 in this case. However, subsequent application of high pressure failed to yield the product 20, but rather gave the tetrazine byproduct 21 and 15. Similar decomposition reactions have been observed previously by Battiste for a bis-7-aza linked pincer material14.
While we have identified a suitable BLOCK containing the desired structural features using molecular modelling, it does not immediately follow that such material will be stable under the coupling procedures necessary to produce molrac materials. Attempted syntheses of the bisepoxide ACE reagent precursor 14 failed to yield the desired material in any quantity but instead yielded several unexpected materials. Similar results were also observed in attempts to utilise the s-tetrazine based coupling protocol.
Since the application of coupling procedures starting with 12 and working outwards have been found to be ineffective, future work must necessarily couple from outside reagents onto 12 (Scheme 5). In particular, the ambient temperature photoACE12 reaction may prove useful as well as s-tetrazine coupling reactions with addition of 12 after dihydropyrazine formation has already been carried out on a second BLOCK.
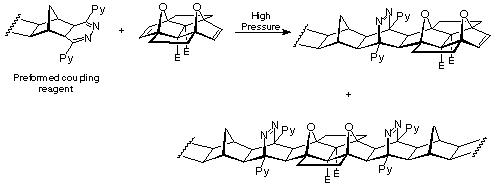
Scheme 5
Molecular modelling was undertaken using Silicon Graphics workstations (O2) and Spartan 5.2 software utilising the AM1 level of theory. Cram’s diene 12 and the relevant precursors were synthesised according to literature procedures10. 1H and 13C NMR spectroscopy was obtained on Bruker AMX300 or Avance400 spectrometers using standard Bruker pulse programs. Spectra are referenced relative to tetramethylsilane.
|
 |
| A solution of
bisfuranocyclophane 10 (1.0 g) and DMAD 11 (3.0 g) in chloroform (2 ml) was
heated overnight in a stainless steel high pressure vessel overnight. The solvent was
removed in vacuo and the residue separated by radial chromatography (petroleum
spirit-ethyl acetate 5:1 to 2:1) to afford 12 (410 mg) and a second product 16
as a yellow coloured solid (400 mg, m.p. 187-189 °C). 1H-NMR (CDCl3): 2.35 - 2.45 (8H, m); 2.59 - 2.83 (4H, m); 2.91 - 3.08 (4H, m); 3.55 (6H, s); 3.80 (6H, m); 6.80 (2H, s). 13 C-NMR (CDCl3): 29.0; 29.4; 1.9; 52.1; 81.2; 97.3; 97.6; 138.9; 147.7; 163.1; 168.8; HRMS calc for C22H22O10: 446.1213, found: 446.1203. |
|
|
 |
| A solution of 12 (0.85 g) DMAD 11
(3.0 g) and RuH2CO(PPh3)3 catalyst (200 mg) in benzene
(20 ml) were refluxed overnight under an nitrogen atmosphere. Purification was achieved by
flash chromatography (silicagel, petroleum spirit - ethyl acetate 3 :1 with increasing
ethyl acetate), followed by radial chromatography (2x) to afford small amounts of 13,
which was recrystallised twice from methanol to afford product as a colourless solid (40
mg, 2.5 %, m.p. 200-202 °C). 1H-NMR (CDCl3): 2.17-2.26 (4H, m); 2.34 - 2.43 (4H, m); 3.55 (4H, s); 3.75 (6H, s); 3.77 (12H, s). 13 C-NMR: 28.9; 47.7; 52.3; 52.6; 76.47; 94.5; 141.23; 161.5; 168.8. HRMS calc for C30H30O14: 614.1638, found: 614.1647. |
|
|
 |
| A mixture of tetraester 16 (200
mg), DMAD 11 (2.0 g) and RuH2CO(PPh3)3 catalyst (200 mg) in benzene (10 ml) was refluxed for 3
days. The reaction mixture was separated by flash chromatography (silicagel, petroleum
ether - ethyl acetate 3 :1, with increasing ethyl acetate), to afford product 17 as
a colourless solid (30 mg, m.p. 191-3 °C) 1H-NMR (CDCl3): 1.80 - 1.90 (2H, m); 2.48 - 2.51 (2H, m); 2.63 - 2.77 (2H, m); 3.18 (2H, s); 3.35 - 3.42 (2H, m); 3.78 (6H, s); 3.79 (6H, s); 4.05 (6H, s). 13 C-NMR (CDCl3): 26.4; 31.5; 51.9; 52.6; 53.0; 53.3; 87.4; 117.2; 139.4; 140.5; 162.4; 163.5; 165.8. HRMS calc for C28H28O14: 588.1479, found: 588.1486. |
|
|
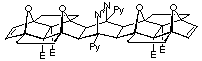 |
| A solution of 12 (50 mg, 0.151
mmol) in DCM (1 ml) was treated with s-tetrazine 18 (36 mg, 0.151 mmol). The
pink color of the tetrazine disappeared within several minutes. The 1H-NMR of crude
mixture revealed that all 12 had been converted to 15 (83%, m.p. 137-9 °C). 1H-NMR (CDCl3) (obtained from crude spectrum): 2.56 - 2.59 (4H, m); 2.81 - 2.84 (4H, m); 3.58 (6H, s); 6.07 (2H, s) 13 C-NMR (CDCl3):30.3, 30.8, 52.3, 109.4, 115.6, 156.7, 163.3, 164.3. HRMS calc for C16H16O6: 304.0947, found: 304.0944. |
|
1. Warrener, R. N. Chem. in Aust., 1992, 59, 578-581.
2. Warrener, R. N.; Johnston, M. R.; Schultz, A. C.; Golic, M.; Houghton, M. A.; Gunter, M. J. Synlett 1998, 590-592.
3. Warrener, R. N.; Ferreira, A. B. B.; Schultz, A. C.; Butler, D. N.; Keene, F. R.; Kelso, L. S. Angew. Chem. Int. Ed. Engl. 1996, 35, 2485-2487.
4. Warrener, R. N.; Schultz, A. C.; Houghton, M. A.; Butler, D. N. Tetrahedron 1997, 53, 3991-4012.
5. Warrener, R. N.; Wang, S.; Russell, R. A.; Gunter, M. J. Synlett 1997, 47-50.
6. Warrener, R. N.; Johnston, M. R.; Gunter, M. J. Synlett 1998, 593-595.
7. Warrener, R. N.; Butler, D. N.; Russell, R. A. Synlett 1998, 566-573.
8. Margetic, D.; Johnston, M. R.; Tiekink, E. R. T.; Warrener, R. N. Tetrahedron Lett. 1998, 39, 5277-5280.
9. Dewar, M. J. S., Zoebisch, E. G., Healy, E. F., J. Am. Chem. Soc., 1985, 107, 3902.
10. Cram, D. J., Knox, G. R., J. Am. Chem. Soc., 1961, 83, 2204, Cram, D. J., Montgomery, C. S., Knox, G. R., J. Am. Chem. Soc., 1966, 88, 515.
11. Warrener, R. N.; Schultz, A. C.; Butler, D. N.; Wang, S.; Mahadevan, I. B.; Russell, R. A. Chem. Commun. 1997, 1023-1024.
12. Margetic, D., Warrener, R. N. unpublished results.
13. Warrener, R. N.; Margetic, D.; Russell, R. A. Synlett 1998, 585-587.
14. Visnick, M., Battiste, M. A., J. Chem. Soc. Chem. Commun., 1985, 1621-622.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0016 as the message subject of your e-mail.