[A0018]
Synthesis of Non-Racemic 1-Hydroxy Cycloalkenecarboxylic Acids by Metathesis of Dialkylated Glycolate Derivatives
Liliana Parra Rapado, Vajira Bulugahapitiya and Philippe Renaud*
Université de Fribourg, Institut de Chimie Organique, Pérolles, CH-1700 Fribourg, Switzerland
E-mail: [email protected]
Received: 31 July 1999 / Uploaded: 12 August 1999
Abstract. A particularly flexible general way to synthesize 1-hydroxy cycloalkenecarboxylic acid derivatives from 2-tert-butyl-2-methyl-1,3-dioxolan-5-one, a chiral equivalent of glycolic acid, is reported. The method is based on a double enolate alkylation of the glycolate derivative followed by ring closing metathesis. A formal synthesis of (-)-quinic acid is reported to demonstate the potential of this approach.
During the recent years, ring closing metathesis has emerged as an extremely useful tool for the synthesis of cyclic compounds of various sizes [1-5]. Recently, we have reported the preparation of the enantiomerically pure dioxolanone 1, a chiral equivalent of glycolic acid [6]. Based on this chemistry, we report here the preparation of 1-hydroxylated cycloalkenecarboxylic acids by use of a sequence of enolate alkylation and olefin metathesis (footnotes 1 and 2). The formal synthesis of (-)-quinic acid is reported to illustrate the potential of the method.
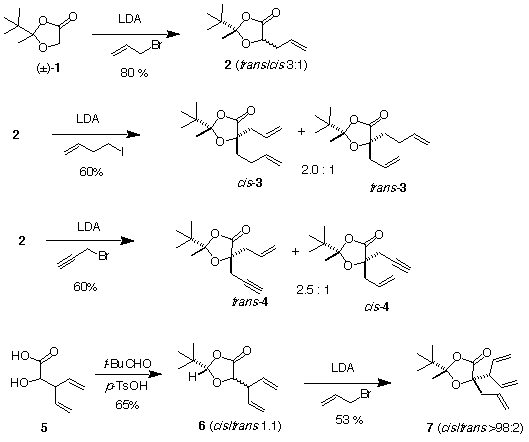
The preparation of dioxolanones 3 and 4 was performed via double alkylation of the chiral glycolate derivatives 1 (Scheme 1).
Scheme 1
The dioxolanone 1 was was first allylated by treatment with LDA and allylbromide to afford 2 as a trans/cis 3.1 mixture of diastereomers (footnotes 3 and 4). When 2 was deprotonated with LDA and alkylated with 4-iodobut-1-ene or propargyl bromide, the alkylated products 3 and 4 were obtained as trans/cis 2:1 and 2.5:1 mixture of diastereomers, respectively. Attempts to use other bases (KHMDS and NaHMDS) and cosolvents (HMPA and DMPU) did not bring any enhancement of the stereoselectivity. This reaction merits some comments since the stereoselectivity of the second alkylation was not as high as expected. Indeed, it has been reported by Seebach that alkylation of 2-tert-butyl-4-methyldioxolanone occurs with very high stereoselectivity [12-15] and we have confirmed this observation in the 2-tert-butyl-2,4-dimethyldioxolanone series [6]. The modest stereochemical control observed here was attributed to allylic 1,3-strain (A1,3 strain) effect at the enolate level (Figure 1). Indeed, in order to avoid repulsion with the tert-butyl group and to minimized A1,3 strain, the vinyl moiety of the propenyl substituent is adopting the configuration depicted in model A, the vinyl group is anti to the tert-butyl substituent and one of the two diastereotopic hydrogen atoms is coplanar with the enolate moiety. The stereochemical outcome to the reaction is then controlled by the antagonist steric effect of the tert-butyl and the vinyl groups. Related effects have been reported in enolate alkylations [16-21] and radical reactions [22]. Kellogs has also observed a drop of selectivity during the alkylation of N,S-acetals from alpha-alkyl-alpha-thiocarboxylic acids when the alpha-alkyl group is a benzyl group, however, this observation was let unexplained [23]. This model was confirmed during the synthesis of 7. Indeed compound 6, prepared by acetalization of the corresponding alpha-hydroxy acid 5 with pinacolone, was allylated in a totally stereoselective fashion (footnote 5). This result is expected from the model discussed above since the enolate derived from 6 should adopt the conformation depicted in model B (Figure 1). The two vinyl goups shield both faces of the enolate in a similar way. Therefore, the stereoselectivity is exclusively controlled by the acetal center (alkylation anti to the tert-butyl group, ul topicity) as in Seebach's case of lactic acid [14].
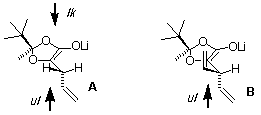
Figure 1. Stereochemical model for the alkylation of Li-enolates derived from 2 (A) and 6 (B): effect of the adjacent prochiral center.
The three compounds 3, 4 and 7 were then treated with Grubbs catalyst 8 [2] to afford the spirocyclic derivatives 9-11 (Scheme 2). The cyclohexene derivative 9 was obtained in 90% yield when the reaction was run with 2 mol% of catalyst at room temperature in benzene. As expected, a cis/trans 2.0:1 mixture of isomers since cis/trans 3 was used. Surprisingly, 10 was isolated in 65% yield as a single diastereoisomer when the reaction was performed in refluxing dichloromethane with 10 mol% catalyst with a trans/cis (2.5:1) mixture of 4 (footnote 6). The minor cis isomer has been lost during this process by decomposition. Finally, the substrate 7 gave the vinylcyclopentene derivative 11 as a 60:40 mixture of diastereomers.
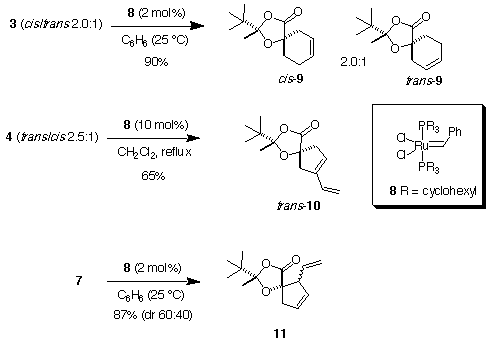
Scheme 2
The utility of this approach was demonstrated by a formal synthesis of (-)-quinic acid (Scheme 3), a ubiquitus natural product playing an important regulating role in the shikimate pathway (footnote 7). Double alkylation of (R)-1 (80% ee) gave the diene (2R,4S)-3 in 51% yield after separation of the diastereomer by chromatography. The diene (2R,4S)-3 afforded diastereomerically pure (2R,4S)-9 in 94% yield upon treatment with 2 mol% of 8. Treatment of (2R,4S)-9 with methanolic HCl gave the ester 12 which was saponified and treated with bromine to afford the product of bromolactonization 13 (89% ee after one recrystallization). Conversion of 13 to (-)-quinic acid has already been reported in the literature [30].
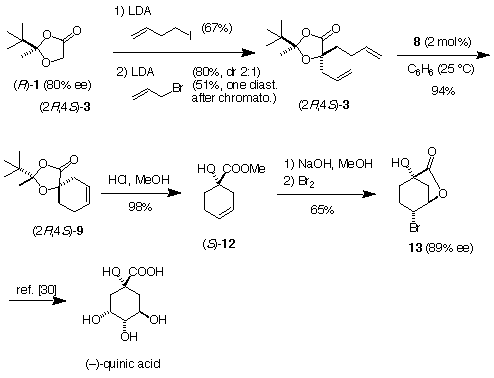
Scheme 3
In conclusion, we have presented here an efficient synthesis of cyclic non-racemic 1-hydroxycycloalkenecarboxylic acid derivatives. The absolute configuration of the alpha-center can be fixed by the choice ot the starting enantiomer of l or by the order of the alkylation sequence. This approach is particularly flexible and should be applicable for a wide range of biologically relevant compounds.
Acknowledgment. This work was supported by the Swiss National Science Foundation. V.B. was supported by a Stipend of the Swiss Confederation.
References
[1] R.H. Grubbs, S.J. Miller, G.C. Fu, Account Chem. Res. 1995, 28, 446.
[2] R.H. Grubbs, S. Chang, Tetrahedron 1998, 54, 4413.
[3] M. Schuster, S. Blechert, Angew. Chem. Int. Ed. 1997, 36, 2037.
[4] S.K. Armstrong, J. Chem. Soc. Perkin Trans. 1 1998, 371.
[5] A. Fürstner, Top. Catal. 1997, 4, 285.
[6] P. Renaud, S. Abazi, Helv. Chim. Acta 1996, 79, 1696.
[7] S. Abazi, L. Parra Rapado, K. Schenk, P. Renaud, Eur. J. Org. Chem. 1999, 477.
[8] K. Hammer, K. Undheim, Tetrahedron 1997, 53, 2309.
[9] K. Hammer, K. Undheim, Tetrahedron 1997, 53, 5925.
[10] K. Hammer, K. Undheim, Tetrahedron 1997, 53, 10603.
[11] S. Khota, N. Sreenivasachary, E. Brahmachary, Tetrahedron Lett. 1998, 39, 2805.
[12] For a comprehensive review, see: D. Seebach, A.R. Sting, M. Hoffmann, Angew. Chem. Int. Ed. 1996, 35, 2708.
[13] D. Seebach, R. Naef, Helv. Chim. Acta 1981, 64, 2704.
[14] D. Seebach, R. Naef, G. Calderari, Tetrahedron 1984, 40, 1313.
[15] G. Frater, U. Müller, W. Günther, Tetrahedron Lett. 1981, 22, 4221.
[16] K. Tomioka, K. Yasuda, H. Kawasaki, K. Koga, Tetrahedron Lett. 1986, 27, 3247.
[17] K. Tomioka, H. Kawasaki, K. Yasuda, K. Koga, J. Am. Chem. Soc. 1988, 110, 3597.
[18] W. Amberg, D. Seebach, Chem. Ber. 1990, 123, 2413.
[19] W. Amberg, D. Seebach, Chem. Ber. 1990, 123, 2429.
[20] W. Amberg, D. Seebach, Chem. Ber. 1990, 123, 2439.
[21] T. Pietzonka, D. Seebach, Chem. Ber. 1991, 124, 1837.
[22] B. Giese, W. Damm, T. Witzel, H.G. Zeitz, Tetrahedron Lett 1993, 34, 7053.
[23] B. Strijtveen, M. Kellog, Tetrahedron 1987, 43, 5039.
[24] T.J. Katz, T.M. Sivavec, J. Am. Chem. Soc. 1985, 107, 737.
[25] N. Chatani, T. Morimoto, T. Muto, S. Murai, J. Am. Chem. Soc. 1994, 116, 6049.
[26] A. Kinoshita, M. Mori, Synlett 1994, 1020.
[27] S.-H. Kim, N. Bowden, R.H. Grubbs, J. Am. Chem. Soc. 1994, 116, 10801.
[28] R. Grewe, W. Lorenzen, L. Vining, Chem. Ber. 1954, 87, 793.
[29] E.E. Smissman, M.A. Oxman, J. Am. Chem. Soc. 1963, 85, 2184.
[30] J. Wolinsky, R. Novak, R. Vasilev, J. Org. Chem. 1964, 29, 3596.
[31] H.J. Bestmann, H.A. Heid, Angew. Chem., Int. Ed. Engl. 1971, 10, 336.
[32] R.M. Meier, C. Tamm, Helv. Chim. Acta 1991, 74, 807.
[33] H. Suemune, K. Matsuno, M. Uchida, K. Sakai, Tetrahedron: Asymmetry 1992, 3, 297.
[34] K.M. Draths, T.L. Ward, J.W. Frost, J. Am. Chem. Soc. 1992, 114, 9725.
[35] K.M. Draths, D.R. Knop, J.W. Frost, J. Am. Chem. Soc. 1999, 121, 1603.
[36] K. Hiroya, K. Ogasawara, Chem. Commun. 1998, 2033.
[37] A. Barco, S. Benetti, C. De Risi, P. Marchetti, G.P. Pollini, V. Zanirato, Tetrahedron: Asymmetry 1997, 8, 3515.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0018 as the message subject of your e-mail.