[B0004]
New Supports for Solid-Phase Organic Synthesis: Development of Cross-Linked Polytetrahydrofuran-Polystyrene Resins
Patrick H. Toy,a Patrick Garibay,b Thomas Hoeg-Jensenb and Kim D. Jandaa,*
a
Department of Chemistry, The Scripps Research Institute and The Skaggs Institute for Chemical Biology, La Jolla, CA 92037 (USA).bNovo Nordisk A/S, Novo Alle 6BS.58, DK-2800 Bagsvaerd (Denmark)
Received: 10 August 1999 / Uploaded: 10 August 1999A manuscript describing the resins presented is currently in press (Tetrahedron Letters) and a manuscript describing the parallel synthesis of a library of phthalide compounds using these resins is also in press (Synlett).
With the advent of combinatorial chemistry and automated synthesis there has been renewed interest in polymer-supported reactions.[1] However, it is evident from the literature that polymer supports used for other than peptide or nucleotide synthesis are, at present, far from optimized. It is not a trivial matter to identify new supports that are economically viable, exhibit satisfactory physical characteristics and that are inert to the diverse range of reagents/catalysts commonly involved in a multi-step combinatorial synthesis. Herein we describe a new class of resins that meet the criteria stated (vide supra). The resins are based on a set of polytetrahydrofuran (PTHF) cross-linkers and hold significant promise as polymer supports for solid-phase synthesis and combinatorial chemistry.
The typical supports used for peptide, small molecule and multi-stage combinatorial synthesis, are lightly cross-linked polystyrene beads with novel linker functionalities that allow for easy detachment of the synthesized compounds. Significantly different supports are those containing grafts of hydrophilic polyethyleneglycol (PEG) on hydrophobic polystyrene beads that were designed for peptide synthesis.[2] The use of bifunctional styrene derivatized PEG chains to cross-link polystyrene as a means for improving general resin performance has also been disclosed.[3] Improved swelling and mechanical properties have been observed with these resins. However, the choice of PEG based cross-linkers can preclude the use of strong bases and organometallic reagents, and imparts hydrophilic properties not always conducive to organic synthesis. To circumvent the inherent problems associated with PEG, an alternative flexible PTHF cross-linker could be installed. Such matrices in principle would provide superior networks to common polystyrene or PEG-polystyrene supports. Herein we report the preparation, characterization and utilization of such PTHF cross-linked polystyrene resins.
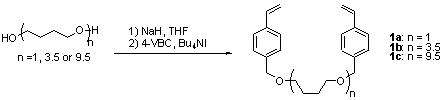
The cross-linkers 1a-c were prepared in 70-85% yield from three diols and 4-vinylbenzyl chloride (4-VBC) using Williamson ether synthesis conditions (Scheme 1).[4] Cross-linker 1a is the product from 1,4-butanediol (mw 90) and cross-linkers 1b and 1c are the products from commercially available PTHF (mw 250) and PTHF (mw 650), respectively. Compounds 1b and 1c were isolated as mixtures of oligomers. It was determined by 1H NMR analysis that the ratios of the oligomers in 1b and 1c were unchanged from the starting diol mixtures. The three different molecular weight diols were used to study possible chain-length dependant swelling and chemical properties.[3f]
Scheme 1. Synthesis of cross-linking agents 1a-1c.
Resins 2a-c were prepared by suspension polymerization of mixtures of 4-VBC, styrene and 1a-c, respectively.[3f,5] Cross-linkers 1a-c were used in 1, 2, 5 and 10 mole percent of the monofunctional monomers to afford 12 resins (Table 1).
The degree to which a resin absorbs a particular solvent and swells is considered to be a good measure of site accessibility and thus resin functionalization.[6] Table 1 compares the swelling of 2a-c to three commonly used supports of the same bead size (100-200 mesh) and approximately the same loading level (ca. 0.9 mmol/g) in solvents typically used for organic synthesis. From the table, it is clear that resins 2a-c have superior swelling properties in all solvents where swelling was observed. As expected, the amount of swelling decreases as the level of cross-linking increases, although even the 10% cross-linked resins swell significantly. It is also apparent that swelling is not dependent on the chain length of the cross-linker. Finally, a series of unfunctionalized resins analogous to 2a-c were prepared by copolymerizing styrene and 1a-c. These beads were found to be stable to a variety of common chemical reagents (> 20 mmol reagent/g resin at rt for 4 hr): mCPBA, Dibal-H, MeI, Ac2O, aq. NaOH, aq. HCl, 50% TFA/CH2Cl2. The only reagents found to degrade the structural integrity of the resins were TMSOTf and BuLi. Importantly, the 5% and 10% cross-linked resins did not degraded upon treatment with BuLi as did the 1% and 2% cross-linked resins.
Table 1. Volumes of swollen resins
Resin - Cross-linking (%) |
Volume of Swollen Resin (mL/g)[a] |
||||
Dioxane |
THF |
DMF |
Benzene |
CH2Cl2 |
|
| Merrifield-1[b] | 6.0 |
6.4 |
4.8 |
6.6 |
6.0 |
| Merrifield-2[b] | 5.4 |
5.4 |
4.2 |
6.6 |
5.8 |
| Tentagel HMP-1[b] | 4.8 |
4.4 |
4.4 |
4.0 |
5.8 |
| 2a-1 | 11.8 |
13.4 |
8.8 |
12.6 |
11.8 |
| 2a-2 | 7.0 |
7.0 |
5.0 |
7.6 |
6.6 |
| 2a-5 | 5.2 |
5.6 |
3.8 |
5.8 |
4.8 |
| 2a-10 | 4.2 |
4.2 |
3.2 |
4.8 |
4.4 |
| 2b-1 | 10.4 |
10.4 |
8.0 |
12.2 |
10.6 |
| 2b-2 | 7.4 |
8.2 |
5.6 |
9.6 |
7.8 |
| 2b-5 | 5.6 |
5.8 |
3.6 |
7.2 |
5.8 |
| 2b-10 | 4.0 |
4.0 |
3.0 |
5.0 |
4.6 |
| 2c-1 | 12.6 |
12.6 |
8.2 |
13.8 |
12.4 |
| 2c-2 | 8.4 |
8.4 |
5.4 |
8.0 |
7.8 |
| 2c-5 | 5.4 |
5.8 |
3.6 |
5.8 |
5.6 |
| 2c-10 | 5.6 |
5.4 |
3.2 |
5.8 |
5.6 |
[a] Volumes were measured in syringes equipped with a sintered frit after equilibrating for 1 h. The resins exhibited modest swelling in ethyl ether and 1:1 THF:water. Water, acetonitrile, and ethanol were also examined, but no swelling was observed. All resins had dry volumes of approximately 1.5 mL/g. [b] Resin was purchased from NovaBiochem, catalog numbers 01-64-0008, 01-64-0104 and 01-64-0106, and used as received.
To assess the synthetic utility of the resins we chose to synthesize phthalide 3[7] via a directed ortho-lithiation approach (Scheme 2).[8] The synthesis of 3 has several important features: (1) A demanding ortho-lithiation reaction is utilized. (2) The trapping step is amenable to accepting a variety of electrophiles. (3) Release of the product via cyclization leaves no "trace" of the linker. (4) To our knowledge this reaction has not previously been performed with benzamides on a polymer support. Thus, N-(4-vinylbenzyl)-phthalimide (4) was synthesized[9] for use in the preparation of resins 5a-c. The aforementioned studies with unfunctionalized polystyrene resins containing 1a-c indicated that 5-10% cross-linking was required to ensure the stability of the resin to BuLi. Therefore, resins 5a-c were prepared from 1a-c with 5% cross-linking by suspension polymerization, substituting 4 for 4-VBC. Resins 5a-c were deprotected with hydrazine and converted to benzamides 6a-c. Treatment of 6a-c with sec-BuLi and TMEDA was followed by trapping with p-anisaldehyde and cyclization to release phthalide 3 in greater than 95% purity, as determined by 1H NMR spectroscopy (Table 2). For comparison, the synthesis of 3 was performed in solution, on divinylbenzene (DVB) and tetraethylene glycol (TEG)[3f] cross-linked polystyrene, and Tentagel resins. It is clearly evident from Table 2 that resins 5a-c afforded higher yields of 3 than did the solution reaction, the two commercially available amine resins and the TEG cross-linked (Table 2, note e) resin studied. It is important to note that the polymer-supported synthesis of 3 has the distinct advantage over the solution synthesis that only the desired product cyclizes and thus cleaves itself from the resin to give essentially pure product. To test the generality of this procedure, phthalides 7-10 (Figure 1) were also prepared using the corresponding acid chlorides and aldehydes. The cleaved products were again determined to be greater than 95% pure as judged by 1H NMR spectroscopy.
Solid-phase organic synthesis has traditionally utilized supports developed for biopolymer synthesis that contain rigid cross-linking agents. The results presented indicate that resins designed specifically for organic synthesis, containing flexible cross-linking agents, may be prepared inexpensively and can provide excellent physical and chemical properties.
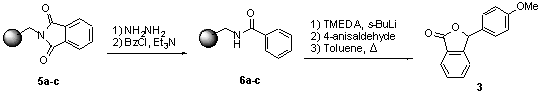
Scheme 2. Resin supported synthesis of 3.
Table 2. Synthesis of phthalide 3.
Resin[a] |
Loading[b] (mmol/g) |
yield[c] (%) |
| 5a | 0.76 |
37 |
| 5b | 0.73 |
34 |
| 5c | 0.68[d] |
43 |
| TEG-NH2[e] | 0.96 |
19 |
| Polystyrene-NH2[f] | 1.25 |
21 |
| Tentagel-NH2[f] | 0.43 |
3[g] |
| Solution | 14 |
[a] All resins prepared for this study contained 5 mol percent of the cross-linker [b] Loading was determined by a FMOC release assay.[10] [c] Refers to yields of 3 that were greater than 95% pure as determined by 1H NMR. [d] A similar loading level was also observed by elemental analysis. [e] The cross-linker for this resin was prepared from TEG and 4-VBC according to the method described in Scheme 1.[3f] An attempt to use this cross-linker in 2 mol percent resulted in degradation of the resin. [f] Resin was purchased from NovaBiochem, catalog numbers 01-64-0094 and 01-64-0143, and used as received. [g] Product was contaminated with PEG chains.
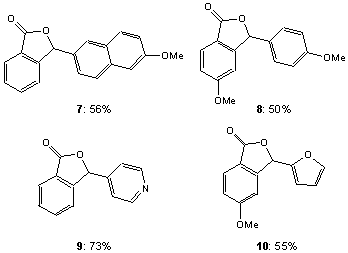
Figure 1. Synthesized phthalides 7-10.
Experimental Section:
Resin 2c-2%: A solution of acacia gum (18.0 g) and NaCl (11.25 g) in water (450 mL) was placed in a 500 mL flanged reaction vessel equipped with a floating magnetic stirrer[3f] and deoxygenated by purging with N2. A solution of VBC (4.59 g, 30.0 mmol), styrene (23.4 mL, 204 mmol), 1c (4.14 g, 4.69 mmol), benzoyl peroxide (0.45 g) in chlorobenzene (30 mL) was injected via pipet into the rapidly stirred aqueous solution. This mixture was heated to 85 ° C for 16 h. The crude polymer was collected and washed in a Soxhlet extractor with water, THF and hexanes. The beads (25.92 g, 86%) were dried in vacuo and sieved to afford resins in 3 size ranges: 50-100 mesh (11.31 g, 38%), 100-200 mesh (10.51 g, 35%) and 200-400 mesh (1.31 g, 4%). MAS 1H NMR (600 MHz, 4 mg per mL CDCL3, 4 s relaxation delay with water suppressed by using a 0.5 s pre-saturation pulse during the relaxation delay): d = 3.75, 1.82. No aromatic resonances are observed.
3, 7-10: Before each reaction, all resins were dried in vacuo (< 0.5 mm Hg, rt) for at least 16 h. Resins 5a-c were refluxed in EtOH/H2NNH2.H2O (20:1) for 16 h. The resin was washed with hot EtOH, H2O, DMF, dioxane, and MeOH, then dried in vacuo. The reaction was judged to be complete by IR spectroscopy. The resins were swollen in dry THF and treated with Et3N (5 eq.) and BzCl (5 eq.). After 1 h the resins were washed with THF, DMF, MeOH and ether, and dried in vacuo to afford resins 6a-c. The reaction was judged to be complete by a negative Kaiser test. Resins 6a-c were swollen in dry THF, cooled to 0 ° C and TMEDA (7 eq.) and sec-BuLi (7 eq.) were added sequentially. After 5 min, p-anisaldehyde (10 eq.) was added. After an additional 30 min the reaction mixture was allowed to warm to room temperature and stirred for 1 h more. The resins were washed as for 6a-c, dried in vacuo, and refluxed in toluene for 16 h. The resins were filtered off and the filtrate was concentrated in vacuo to afford 3. 1H NMR (300 MHz, CDCl3): 7.97 (d, 1 H, J = 7.5 Hz), 7.69 (t, 1 H, J = 7.5 Hz), 7.56 (t, 1 H, J = 7.5 Hz), 7.32 (d, 1 H, J = 7.5 Hz), 7.18 (d, 2 H, J = 8.8 Hz), 6.89 (d, 2 H, J = 8.8 Hz), 6.38 (s, 1 H), 3.81 (s, 3H). To mirror the solid-phase synthesis, the solution synthesis of 3 utilized benzyl benzamide as the lithiation substrate and the product required chromatographic purification. The synthesis of 3 on TEG cross-linked polystyrene used resin prepared in the same manner as were 5a-c. The synthesis of 3 on DVB and Tentagel resins did not require deprotection of the amine prior to benzamide formation. Phthalides 7-10 were prepared by similar methods on 5% cross-linked 5c using the appropriate acid chlorides and aldehydes with the exception that lithiation was performed with n-BuLi in the absence of TMEDA.
References:
[1] a) J. S. Fruchtel, G. Jung, Angew. Chem. 1996, 108, 19; Angew. Chem. Int. Ed. 1996, 35, 17; b) L. A. S. Booth, P. H. H. Hermkens, H. C. J. Ottenheijm, D. Rees, Tetrahedron 1998, 54, 15385; d) R. C. D. Brown, J. Chem. Soc. Perkin Trans. 1 1998, 3293.
[2] a) E. Bayer, Angew. Chem. 1991, 103, 117; Angew. Chem. Int. Ed. Engl. 1991, 30, 113; b) O. W. Gooding, S. Baudart, T. L. Deegan, K. Heisler, J. W. Labadie, W. S. Newcomb, J. A. Porco Jr., P. v. Eikeren, J. Comb. Chem. 1999, 1, 113; c) S. A. Kates, B. F. McGuinness, C. Blackburn, G. W. Griffin, N. A. Sole, G. Barany, F. Albericio, Biopolymers 1998, 47, 365.
[3] a) S. Itsuno, I. Moue, K. Ito, Polym. Bull. 1989, 21, 365; b) S. Itsuno, Y. Sakurai, K. Ito, T. Maruyama, S. Nakahanma, J. M. J. Frechet, J. Org. Chem. 1990, 55, 304; c) K. Kamahori, K. Ito, S. Itsuno, J. Org. Chem. 1996, 61, 8321; d) M. Renil, M. Meldal, Tetrahedron Lett. 1996, 34, 6185; e) J. Buchardt, M. Meldal, Tetrahedron Lett. 1998, 39, 8695; f) M. E. Wilson, K. Paech, W. J. Zhou, M. J. Kurth, J. Org. Chem. 1998, 63, 5094.
[4] C. Bougherara, B. Boutevin, J. J. Robin, Polym. Bull. 1991, 26, 181.
[5] D. C. Sherrington, Chem Commun. 1998, 2275.
[6] R. Santini, M. C. Griffith, M. Qi, Tetrahedron Lett. 1998, 39, 8951.
[7] M. Iwao, H. Inoue, T. Kuraishi, Chem. Lett. 1984, 12S63.
[8] a) V. Snieckus, Chem. Rev. 1990, 20, 879; b) S. Havez, M. Begtrup, P. Vedso, K. Anderson, T. Ruhland, J. Org. Chem. 1998, 63, 7418.
[9] H. W. Gibson, F. C. Bailey, Macromolecules 1977, 10, 602.
[10] M. Hori, D. J. Gravert, P. Wentworth Jr., K. D. Janda, Bioorg. Med. Chem. Lett. 1998, 8, 2363.
This work was supported financially by the National Institutes of Health (GM-56154), the Scripps Research Institute and the Skaggs Institute for Chemical Biology. MAS NMR spectra were recorded at Hoechst Marion Roussel by Dr. J. A. Malikayil. We thank BASF AG for the gift of polytetrahydrofuran 250.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with B0004 as the message subject of your e-mail.