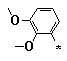
[B0005]
Symmetric Building Blocks and Combinatorial Functional Group Transformation as Versatile Strategies in Combinatorial Chemistry
Jörg Steffen Früchtel, Karin Pflugseder, and Hubert Gstach*
Novartis Forschungsinstitut Wien, Brunnerstrasse 59, A-1235 Vienna, Austria
Received: 10 August 1999 / Uploaded: 11 August 1999
Abstract
A representative set of symmetric diacids was coupled onto deprotected TentaGel Rink Amide resin. The symmetric building blocks served as model to complete a synthesis protocol and to switch to a different synthesis paradigm consecutively. The reaction sequence continued in a non-combinatorial step by coupling of a bifunctional reagent (3-aminoacetophenone) to the remaining carboxyfunction of the symmetric diacid. The ketone served as model of a reagent prepared for combinatorial functional group transformation. The arylmethylketone was reacted with a set of aryl- and heteroarylaldehydes to give a,b-unsaturated ketones. Subsequently, guanidine, alkyl-, and arylcarboxamidines were introduced in combinatorial synthesis of substituted pyrimidines by reaction with the a,b-unsaturated ketone functionality. The combination of symmetric building blocks and combinatorial functional group transformation created a versatile reaction sequence ideally suited for production of libraries from libraries with added diversity.
Key words: symmetric building blocks; combinatorial functional group transformation; libraries from libraries; Claisen-Schmidt reaction; a,b-unsaturated ketones; chalcones; pyrimidine synthesis.
Introduction
An impressive repertoire of synthetic procedures of organic chemistry has been adapted and optimized to work reliable on various solid supports.1 The currently available experimental protocols enable the synthesis of large numbers of compounds (individual chemical entities as well as libraries of compounds) by techniques of combinatorial chemistry.2 The rapidly growing knowledge in combinatorial chemistry has developed to an integral part of the modern drug discovery process in pharmaceutical industry.3
The most time consuming step in synthesis of compounds on solid support is the extensive reaction development to optimize yields and purity of the envisaged products. The final production process of a library is fast compared to the planning and development phase. Most of the efforts must be invested in exploring the scope of reactions, testing of building blocks, optimizing reaction conditions, and final design of the library taking in account all the primary results generated throughout the reaction development. Therefore, it is highly desirable to look for combinatorial chemistry strategies to smooth the balance between initial and final phase of the overall process. An attractive strategy to solve the problem is to make repeated use of chemical procedures already worked out, and tested in a different experimental context of a previous synthesis. Such a strategy, “functional group transformation”, was proposed by Hermkens & Hamersma, recently.4 In this efficacy-enhancing concept a library produced in a primary reaction sequence serves as starting point for a second-generation library prepared with most of the building blocks, and synthetic procedures tested for the first one. Key is the introduction of a suitable functional group that can be transformed to a key intermediate to prepare the second-generation library from the first library. The authors have shown by computational methods that such transformed libraries contain as much new structural diversity as an entirely new created library. Other approaches to maximize the return on invested synthetic efforts have been described, like concepts “libraries from libraries” or “postsynthesis modification”, respectively.5
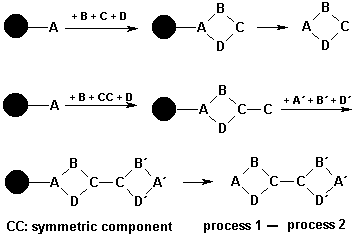
Another powerful strategy to which we have focused our study is linking of chemical processes by symmetric building blocks. The concept is exemplified by comparison of a multi component reaction (MCR), without and with a symmetry containing component (scheme below).
After the first reaction cycle a symmetric component employed in a MCR provides a functional group prepared to perform of a second cycle. The integration of MCRs to linked MCRs has become an interesting aspect of combinatorial chemistry. Successful examples are reported.6 Further approaches to improve the productivity of MCRs were made by introduction of “universal building blocks” that can be converted to new functionalities.7 The concepts discussed above apply proven synthetic procedures. The accessible diversity of products is nearly unlimited. However, unions of MCRs very likely generate molecules with exceptional high molecular weight. The bioavailability and pharmacokinetics of such products may be unfavourable.8
During the last decades a couple of different classes of unprotected symmetric reagents have been immobilized on solid supports, spanning over a broad range of functional groups like diamines, dialdehydes, diketones, diacid chlorides, diacids, or diisocyanates, respectively.9 The main focus was to distinguish identical functional groups of symmetric reagents. By attachment of the reagent to the solid support the reamaining functionality was available for synthesis of unsymmetrically substituted derivatives. The reagent was seen more as a scaffold rather than a building block. A versatile application of symmetric reagents was demonstrated by synthesis of an ergoline library.10 Cyclohexanedioic acid and symmetric diamines were introduced as building blocks to modify a functionalized alkaloid (lysergic acid amine). Symmetric diamines were extensively used in synthesis of structurally heterogeneous combinatorial libraries.11 We report on a model study, where the focus is given to the restart of a combinatorial synthesis that was closed by reaction with unprotected symmetric diacids. In a conceptually different scheme a protected symmetric diacid was used for chemical ligation of one library with another library to demonstrate “double combinatorial chemistry”.12 Mono-allylated 2,6-naphthalene dicarboxylic acid and phthalic anhydride were applied in a combinatorial synthesis of balanol analogues.13 In our approach, the functional group for continuation of synthesis is provided as the unreacted carboxy terminus of a symmetric alkyl, aryl, or heteroaryl diacid, respectively. Subsequently, a bifunctional building block (3-aminoacetophenone) is coupled to the carboxy function in a non-combinatorial step to gain further synthetic flexibility. Finally, the combinatorial potential of the reaction sequence is restored by a combinatorial functional group transformation of the arylmethylketone to a,b-unsaturated ketones following known procedures. These key intermediates are further reacted in a subsequent combinatorial step with guanidine, alkyl- and arylcarboxamidines to yield substituted pyrimidines.
Results and Discussion
We started our feasibility study with immobilization of a representative set of unprotected, symmetric alkyl, aryl, and heteroaryl diacids (Figure 2) on TentaGel Rink Amide 1 (see Figure 1).14 The coupling of the diacids to the deprotected amino group of the linker was easily achieved by reacting the resin with a 0.2 M solution of the corresponding, preactivated symmetric acid following standard amide coupling protocols.15 1-hydroxybenzotriazole (HOBt) and N,N´-diisopropylcarbodiimide (DIC) were used as coupling reagents to give resins 2a-d. The completion of the reaction was checked by a Bromophenol Blue test.16 A ten fold excess of the preactivated acids was applied to minimize symmetric amide formation by site interactions in the solid support. The analysis of the crude products (6) did not reveal significant by-products which would have been indicative for site interactions during the first step of the reaction. The results confirmed recent studies on site interactions in resin supports performed with symmetric diacid chlorides.17 The next step in the synthesis was the coupling of 3-aminoacetophenone to the unprotected carboxy function provided by the immobilized symmetric diacids on resins 2a-d to prepare for the planned switch to combinatorial functional group transformation. Resins 2a-d were treated with a 0.23 M solution of 3-aminoacetophenone, HOBt, and DIC, respectively to result in 3a-d. Subsequently, the resins 3a-d were subjected to Claisen-Schmidt reaction with a set of aromatic and heteroaromatic aldehydes to give resins 4a-d. The Claisen-Schmidt reaction of immobilized aldehydes and acetophenones has been studied by Marzinzik & Felder18 in detail. The resulting chalcones were shown to be versatile intermediates for synthesis of divers heterocycles. A Claisen-Schmidt reaction with an immobilized symmetric aldehyde was also successful.9c The scope of the reaction demonstrated the high value of a,b-unsaturated ketones as key intermediates in combinatorial chemistry. The diversity and synthetic flexibility accessible in reactions of a,b-unsaturated ketones establishes an ideal tool in the concept of functional group transformation. Only a few examples of Claisen-Schmidt reactions are described where the acetophenone derivative is first immobilized and reacted with aldehydes consecutively.19 We had to optimize the reaction conditions for Claisen-Schmidt reaction on TentaGel support, because all described procedures were performed on polystyrene resins. Initial attempts to react the resin bound 3-aminoacetophenones (3a-d) with aromatic aldehydes employing LiOH-DME conditions did not result in satisfactory purities of condensation products. Finally, KOH-MeOH was found to be the base of choice for reaction on TentaGel. Resins 3a-d were treated with KOH (as a 0.7 M solution in MeOH) and a 23-fold excess of benzaldehyde to give resin bound chalcones 4a-d. The a,b-unsaturated ketone functionality contained in 4a-d was utilized for subsequent synthesis of 2-substituted pyrimidines by reaction with an excess of thiophene-2-carboxamidine at 100 oC to give resins 5a-d. The 2-thienyl substituted pyrimidine derivatives 6a-d were obtained after cleavage from the solid support using TFA. The crude products (6a-d) were purified and characterized analytically (NMR, HRMS). To exploit the scope of the Claisen-Schmidt reaction on TentaGel a variety of aromatic aldehydes (Figure 3) were tested under the same reaction conditions as applied for resins 4a-d. In this series the symmetric diacid component (heptanedioic acid) and the nitrogen nucleophile employed (guanidine) in the heterocycle forming step were kept constant. The resin bound 2-aminopyrimidines 5e-k were cleaved (TFA) from the solid support. The final 2-aminopyrimidines 6e-k were characterized analytically. A third series of experiments addressed the scope of carboxamidines applicable for pyrimidine ring closure on TentaGel. The symmetric diacid (heptanedioic acid) as well as the aldehyde component (benzaldehyde) were kept constant, and a representative selection of commercially available carboxamidines (Figure 4) was reacted with resin 4a to give resin-bound 2-alkyl-, and 2-arylsubstituted pyrimidines 5l-v. After cleavage from the resin the products 6l-v were purified and characterized analytically.
A detailed description of the experimental procedures and the analytical data of pyrimidines 6a-v will be published elsewhere. The yields and the purities of 6a-v are summarized in table 1.
Figure 1.

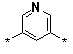
Figure 2. Pyrimidines 6a-d (R1 = a-d, R2 = Ph, R3 = thien-2-yl); diacids
a |
b |
c |
d |
|
|
|
|
Figure 3. Pyrimidines 6e-k (R1 = -(CH2)5-; R2 = e-k; R3 = NH2); aldehydes
e |
f |
g |
h |
i |
j |
k |
|
|
|
|
|
|
|
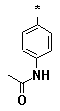
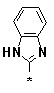
Figure 4. Pyrimidines 6l-v (R1 = -(CH2)5-; R2 = Ph; R3 = l-v); carboxamidines
l |
m |
n |
o |
p |
q |
|
|
||||
r |
s |
t |
u |
v |
|
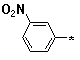 |
|
|
|
Table 1. Yields and MS-analysis of substituted pyrimidines 6a-v
entry |
% yield of crudes$ |
% purity of crudes (254 nm) |
% yield of purified |
MS (ESI) base peak (100%) |
Symmetric diacids |
||||
6a |
99 |
63 |
35 |
471 (100) |
6b |
65 |
65 |
21 |
477 (100) |
6c |
83 |
67 |
24 |
483 (100) |
6d |
42 |
65 |
14 |
478 (100) |
Aldehydes |
||||
6e |
96 |
79 |
33 |
438 (100) |
6f |
102 |
86 |
41 |
434 (100) |
6g |
110 |
77 |
41 |
464 (100) |
6h |
104 |
88 |
40 |
448 (100) |
6i |
100 |
85 |
44 |
461 (100) |
6j |
95 |
76 |
31 |
405 (100) |
6k |
96 |
90 |
31 |
410 (100) |
Carboxamidines |
||||
6l |
62 |
73 |
27 |
451 (100)# |
6m |
62 |
75 |
29 |
467 (100)# |
6n |
70 |
80 |
24 |
545 (100)# |
6o |
98 |
86 |
36 |
487 (100)# |
6p |
75 |
87 |
30 |
501 (100)# |
6q |
103 |
80 |
30 |
522 (100)# |
6r |
82 |
85 |
32 |
555 (100)# |
6s |
76 |
90 |
34 |
532 (100)# |
6t |
83 |
83 |
27 |
488 (100)# |
6u |
76 |
74 |
34 |
n.d§ |
6v |
86 |
56 |
36 |
n.d§ |
$
yields>100% are due to PEG-impurities cleaved from the resin; #as Na-adduct; §confirmed by HRMS.Conclusions
The performed model study on linking of chemical processes validated that a combination of symmetric building blocks and a combinatorial functional group transformation creates a versatile strategy to improve the productivity of available synthetic procedures. The strategy is therefore a powerful tool for production of libraries from libraries with added diversity.
Acknowledgements
We would like to thank Dr. Andrea Steck for structure confirmations by NMR – spectroscopy, and Francis Roll for analysis by HRMS. Arno Pruckner is acknowledged for excellent technical assistance. We are doubtful to Dr. Andreas Marzinzik, Dr. Eduard Felder, and Dr. James Wareing for conceptual discussions.
References and Notes
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with B0005 as the message subject of your e-mail.