[B0006]
Preliminary Results: The Development of a Continuous Flow Injection Microreactor For Organic Synthesis and Combinatorial Applications using Wittig Chemistry
Skelton, V., Greenway, G.M., Haswell, S.J., Morgan, D.O*., Styring, P., Warrington, B.H.**, Wong, S. Y. F. **
Department of Chemistry, Faculty of Science and the Environment,
University of Hull, Cottingham Road, Hull, HU6 7RX. 01482 466409.
E-mail: [email protected]
* SmithKline Beecham Pharmaceuticals, Old Powder Mills, Nr Leigh,
Tonbridge, kent, TN11 9AN
** SmithKline Beecham, New Frontiers Science Park (North), Third Avenue,
Harlow, Essex, CM19 5AW
Received: 10 August 1999 / Uploaded: 11 August 1999
Abstract
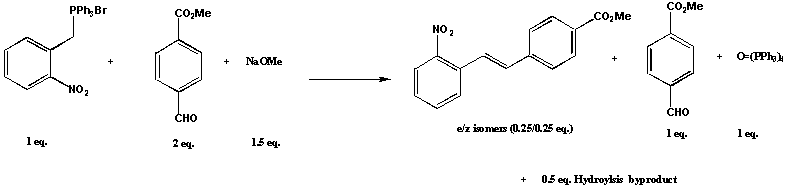
This poster outlines the preliminary data obtained using microreactor technology, which has the potential to offer enhanced reaction optimisation and combinatorial based synthesis and screening. Initial developments have centred on the synthetic application of microreactors to Suzuki [1] and the well-established Wittig synthesis [2]. This particular poster focuses on the Wittig syntheses, Figure 1 in microreactors and will compare the performance characteristics to traditional batch methodology.
The poster outlines the reaction yield data using a continuous flow injection microreactor prepared in borosilicate glass (channel geometries 300 microns wide and 100 microns deep) using photolithographic patterning and modified wet etch techniques [3]. The fabrication method, reaction optimisation and microreactor adaptation, including injection optimisation and reaction stoichiometry alteration (1:1 eq.) are described. The resulting optimised method was used with a variety of aldehydes to demonstrate its general applicability. The work extends into the development of a microreactor that will facilitate combinatorial synthesis and detection using the optimised reaction methodology described earlier, enabling the development of a semi automated combinatorial system with the potential for ultra high throughput operation.
Figure 1: Wittig reaction investigated in the microreactor
Introduction
The microreactor concept was realised by Ponton[4] in 1993, describing a hypothetical miniaturised disposable chemical plant where a parallel scale out approach could produce significant batch quantities of product whilst allowing safe 'at point of use' manufacture of HCN for example. Recently, Chambers [5] reported the selective fluorination of various organic species, however the system described used a nickel microreactor requiring reagent delivery via syringe or syringe pumps. In this study solvent and sample pumping is achieved by electroosmotic flow (EOF) [6] rather than hydrostatic displacement as used by Chambers. EOF has been selected for this particular application as it offers a simpler sample handling methodology with greater control and selectivity of the reaction chemistry.
The work described here is based on the optimisation of a Wittig synthesis which is used in industry for a variety of syntheses e.g. vitamin A producing a C-C link with the formation of olefinic double bond. The reaction was selected for two reasons, firstly to demonstrate synthetic application of microreactors and secondly, to establish the potential to perform combinatorial chemistry in such devices. Previously reported methodology [1] has illustrated the potential of microtechnology to perform catalysed organic reactions. The Wittig synthesis however offers simple solution chemistry, whilst offering the advantage of producing a coloured intermediate (ylide), which allows visual solution profiling and diffusive mixing to be achieved. The following sections will briefly outline the methodology and the preliminary results obtained.
Experimental
The flow injection microreactor was fabricated using photolithographic techniques [3]. Microporous silica frits (MPSF) were generated using the method outlined previously [7] and positioned in microchannels as indicated in figure 2. The channels were then sealed by annealing to the patterned base plate to a 17 mm thick top plate (680 oC) in a microwave furnace. The top plate included 3 mm internal diameter pre-drilled holes aligned at the ends of each channel to act as reservoirs and electrode supports.
Figure 2: Schematic diagram of the 'T'-shaped manifold used in the reactor for the Wittig synthesis
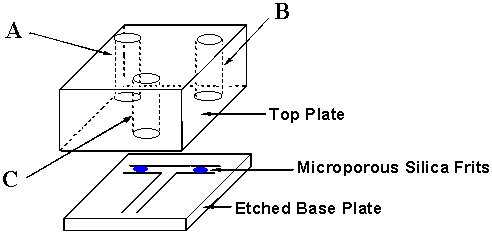
The synthetic method used was adapted from that previously reported by Hughes [8, 9] et al. The continuous flow micro-reactor was operated in the following manor. Prior to synthesis, the microchannels were primed with dry, degassed methanol (MeOH) removing any excess moisture from the channels and reducing the side reaction of hydrolysis. A standard solution of 2-nitrobenzyltriphenylphosphine bromide (80 microlitres, 0.01 M) in MeOH was added into reservoir A of the microreactor. Methyl-4-formylbenzoate (0.02 M) was basified with sodium methoxide (0.3 ml, 0.5M standard solution concentration) and 80 microlitres of the premixed solution was introduced into reservoir B. MeOH (40 microlitres, dry, degassed) was introduced into reservoir C. Electrodes were then placed in each reservoir (A + B positive, C ground), which were then covered using laboratory film. Two methods of mobilisation were employed.
Mobilisation method 1: an external applied voltage (in the range of 100 – 700 V) was applied to channels A (+) and B (+) relative to reservoir C (-). This induced the continuous flow of methyl-4-formylbenzoate from B (+) to C (-) and 2-nitrobenzytriphenylphosphine bromide from A (+) to C (-). This method was used for the conventional batch methodology i.e. the aldehyde in excess for both synthetic and the combinatorial approaches.
Mobilisation method 2: involved the application of the external voltage (400V) to reservoir B (+) relative to C (-), inducing the flow of methyl-4-formylbenzoate. The periodic application of 400V between A (+) to C (-) efficiently injected reagent slugs of 2-nitrotriphenylphosphine bromide into the continuous stream of methyl-4-formylbenzoate. Mobilisation method 2 was used for the synthetic optimisation of 1:1 stoichiometry in the microreactor. The microreactor was run for 20 minutes, after which the reaction was quenched using dilute HCl before analysis. For both of the above methods, the total volume of the solutions in reservoir A and C at the end of each reaction was recorded and samples were taken for HPLC-MS analysis. The LC peak area was used to determine the solution yield of the reaction.
Synthetic Reaction Development
Using mobilisation method 1 the optimum reaction yield relative to the applied voltage or flow was obtained. At high voltage it was apparent that the reaction was not occurring in the reaction manifold (the purple ylide intermediate was observed in reservoir C), which resulted in no increase in yield compared with traditional batch reactions. At lower voltages (ie. less than 400 V), the purple intermediate was observed in the mixing channel of the reactor and the reaction yield was increased by approximately 40% over the batch method, however the yield obtained was non-reproducible and yields of over 100% were obtained (mainly at 100V). The reaction was optimised further at 400V.
Figure 3 summarises the results obtained for the synthetic and combinatorial approach carried out at 400V for both the continuous (Method 1) and injection (Method 2) profiling. The initial section of the graph summarises the results for methyl-4-formylbenzoate. The traditional batch yield using the basified aldehyde was 60% (2:1 stoichiometry), however the microreactor using continuous flow of both reagents was 70% (2:1), demonstrating a 10% increase in the reaction yield. For stoichiometry of 1:1, the batch reaction yield was 48% and the microreactor exhibited an 11% increase. Whilst the yield increase for the microreactor may not seem substantial, the time to perform the experiments was significantly reduced (20 minutes batch, <5 minutes microreactor). This aspect of microreactor technology is very powerful in synthetic chemical application where one may wish to see if a simple reaction is working. The general applicability of the microreactor was tested using four other aldehydes exhibiting different diversities. As can be seen from the remainder of the graph, the 3-benzyloxybenzaldehyde and 2-naphthaldehyde exhibited very similar yields when compared with that of the batch synthesis however, the yield obtained for 5-nitrothiophene-2-carboxaldehyde (1:1) was 9% lower than for the traditional scale. It must be noted that the four aldehydes tested used the optimised method for the methyl-4-formylbenzoate. This approach allows for the development of a combinatorial device allowing screening for a variety of compounds.
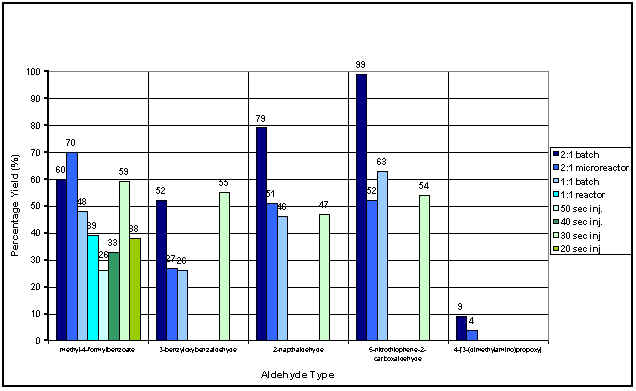
Figure 3: Result Summary for Combinatorial and Synthetic experiments
Combinatorial Chemistry Prototype
Combinatorial chemistry allows the synthesis of a large number of organic species in which a large number of chemical combinations are achieved. The methodology enables the fast generation and screening of combinatorial libraries against biologically active target compounds ideal for the drug discovery process. Combinatorial libraries exhibit greater SAR (structure activity) and therefore increased likelihood of drug discovery success due to the immense libraries that can be synthesised using very few synthetic steps (fifty individual reactions coupled with fifty containers can synthesise a diverse library of 100,000 members in one week). The speed of combinatorial analysis is already at a significant scale however, the attributes of microtechnology offer a highly selective method potentially increasing the number of potential members that could be synthesised per week. The following information describes the development of the optimised synthetic reaction outlined previously enabling the production of a semi-automated prototype combinatorial screening device.
Figure 4 is a schematic of a simple semi-automated screening device. In this case the microreactor manifold is different to that previously described allowing the positioning of the fibre optics leading from the halogen light source to the diode array detector. The reservoir (magnified) shows the positioning of a PTFE block that has been machined to fit the reservoir diameter. The block consists of an inlet and outlet section to which PTFE tubing is connected allowing the delivery/removal of solution/reagents. The correct amount of solution/reagent can be administered into the reservoir by using a flow injection loop that allows the injection of the following profile, aldehyde, methanol, aldehyde, and methanol preventing any cross contamination. The reagents are delivered from each reservoir and react infront of the fibre optic detection system and then flow to waste. New starting materials are introduced from the autosampler located in the flow injection manifold. The reaction and detection can be determined all on-chip however, for the system to be fully optimised, a secondary micro-total analytical system is required allowing product isolation and reagent recycling. The system described in Figure 4 is currently under further investigation.

Figure 4: Schematic of semi-automated aldehyde screening device
Conclusion
Microtechnology offers the potential for novel tools with which to control chemistry and enable unique insight into reaction chemistry to be achieved. For many synthetic chemical applications such devices will offer, for example, increased reaction yields and reduction in stoichiometry. Further the development of a semi-automated combinatorial screening device potentially offers a rapid screening device producing a vast and varied combinatorial library.
Acknowledgements
This work was supported by the EPSRC and CASED by SmithKline Beecham Pharmaceuticals.
References and Notes
[1] G. M. Greenway, S. J. Haswell, D. O. Morgan, V. Skelton and P.Styring submitted to Sensors & Actuators-B
[2] G. M. Greenway, S. J. Haswell, D. O. Morgan, V. Skelton, P.Styring, B.H. Warrington, and S. Y. F. Wong submitted to Sensors andActuators-B
[3] Daykin, R.N.C., Haswell S.J., Anal. Chim. Acta, 1995, 313, 155.
[4] J.W, Ponton, in Microreaction Technology, Proceedings of the First International Conference on Microreaction Technology, (Ed. W. Ehrfield), Springer-Verlag Berlin ]
[5] Chambers, R.D., Spink, R.C.H., Chem. Commun., 1999, 10, 883 - 884
[6] P.D.I. Fletcher, S.J. Haswell, and V.N. Paunov, Analyst, 1999, in press.
[7] P.D. Christensen, S.W.P. Johnson, T. McCreedy, V. Skelton, N.G. Wilson, Anal. Commun., 1998, 35, 341.
[8] I. Hughes, W. P. Nolan, R. A. Raphael, J. Chem. Soc. Perkin Trans., 1990, 1, 2475
[9] I. Hughes, Tett. Lett. 1996, 37(42), 7595
All comments on this poster should be sent by e-mail to (mailto:[email protected]
ona.edu) [email protected] with B0006
as the message subject of your e-mail.