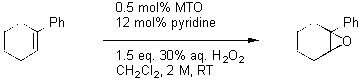
[B0011]
Automation in Reaction Optimization
Jay P. Chiang* and K. Barry Sharpless
The Scripps Research Institute, Department of Chemistry, 10550 N. Torrey Pines Road,
La Jolla, CA 92037
E-mail: [email protected]
Received: 20 August 1999 / Uploaded: 30 August 1999
The use of automation in high-throughput screening and high-speed parallel synthesis has been well documented in recent years1. While combinatorial techniques and automation are beginning to permeate material science and catalyst research2, the benefits of automation in reaction development is a largely untapped.
Our approach towards automating reaction optimization is based on the utilization of a robotics lab designed to accommodate a wide range of applications. This is achieved through the principle of unit operations. Unit operations can be described as basic tasks that are performed normally in chemistry such as the manipulation of liquids and the weighing of starting materials. This approach is based on using a number of workstations that can achieve the automation of these tasks and accelerate a given operation. Automated workstations are designed to carry out a small number of tasks very well. They are most useful when employed as specialized instruments. When they are built to perform complex processes, problems often arise.
A synthetic procedure can be reduced to basic tasks and these individual tasks can be automated. For example, a general route in the synthesis of compounds in solution phase involves making solutions of starting materials, dispensing solutions of starting materials to reaction vessels, heating and agitation, work-up of the products, and analysis. Each of these steps in considered a unit operation and is performed separately on individual platforms. There are many theories in automating chemistry. Other approaches involved the development of robotic workstations capable of performing all the tasks in the sample procedure. Often this results in an inefficient platform that has only modest capabilities in each operation.
Our automated lab has been applied towards the study and optimization of catalytic reactions. A reaction that has recently benefited from the accelerated rate of screening is the methylrhenium trioxide (MTO) -catalyzed epoxidation of olefins using hydrogen peroxide. Herrmann first reported this process using anhydrous conditions3. Our group made a number of improvements on Herrman’s original system that greatly improved the scope4. The addition of pyridine reduced the acidity of the reaction mixture allowing the synthesis of sensitive epoxides. There is some evidence that the pyridine also acts as a phase transfer agent. Our modified epoxidation also employed aqueous hydrogen peroxide and a solvent change from t-butanol to methylene chloride. Our initial reaction conditions call for the use of 0.5% MTO, 12 mol% pyridine, and 1.5 equiv 30% aqueous hydrogen peroxide in methylene chloride (scheme 1).
Scheme 1. Epoxidation of 1-phenylcyclohexene by MTO/pyridine (50 mmol scale).
These initial findings led to an epoxidation process of acid-sensitive epoxides. While epoxides of cis olefins were formed efficiently, some terminal and trans olefins required long reactions times or did not reach full conversion. Examination of the reaction conditions for the pyridine system reveals a large number of variables. We focused on varying the pyridine additive, the oxidant, and the catalyst in order to optimize the epoxidation of terminal and trans alkenes.
The reactions for the screening of the various conditions were performed using a number of automated workstations. Stock solutions of olefin, catalyst, and pyridine were prepared using a weighing and dissolution station (Figure 1). This workstation manufactured by Bohdan Automation is designed for the preparation of stock solutions of reagents.

Figure 1. Bohdan weighing and dissolution station is used to make stock solutions of starting materials and reagents. Fitted with a balance, a vortex, and an arm equipped with cannula and grippers capable of lifting tubes, up to 192 solutions can be made at a time.
The reactions were performed using a Gilson 215 liquid handler. This liquid handler was fitted with two stir plates that could hold four reactions each (Figure 2). Stock solutions of the olefin, pyridine, and catalyst were added and stirred on the deck of the instrument. The oxidant was then added to each reaction vessel in intervals of 30 seconds. This gives the instrument adequate time to take an aliquot and rinse the needle. A timer was started for each reaction once the oxidant was added. Aliquots for each reaction were taken at appropriate timepoints and these timepoints were quenched and dried in test tubes preloaded with catalytic MnO2 and Na2SO4. A reaction profile can be generated for reactions with various reaction times. Generally, the aliquots were spaced such that a reasonable profile could be generated with 12 time points. These aliquots were then diluted to a suitable concentration for GC analysis.

Figure 2. Gilson 215 Liquid Handler is versatile workstation shown here adapted for reaction monitoring. Shown here, the two stir plates hold 4 scintillation vials each but can be adapted to hold a variety of vial sizes. A variety of racks can be placed on the deck of the instrument to accommodate reagent stock solutions, tubes for quenching aliquots, and analytical vials.
Using this reaction-screening platform, 16 reactions could be monitored in a single day (assuming no longer then 12-hour reaction times) in which 12 timepoints were taken per reaction. With a run time of 15 minutes for each GC run, a total of 96 samples can be run in a day per GC. We employed 2 GCs and were able to run 192 samples, which accommodated the 16 reactions, performed each day. In this case the rate of screening coincides with the rate of analysis.
With the large number of reaction conditions screened, a summary of the results for the best conditions will be described. The effect of the pyridine additive and amount added had a dramatic effect on the conversion of various olefins. It was found that the use of 10 mol% 3-cyanopyridine was the most efficient pyridine additive for the conversion of 1-decene to the corresponding epoxide5 (Figure 3). The addition of 3-cyanopyridine greatly improved the conversion when compared to the reaction with no additive, which did not reach full conversion. It is interesting to note that the reaction with the pyridine additive actually fared worse then the blank reaction.
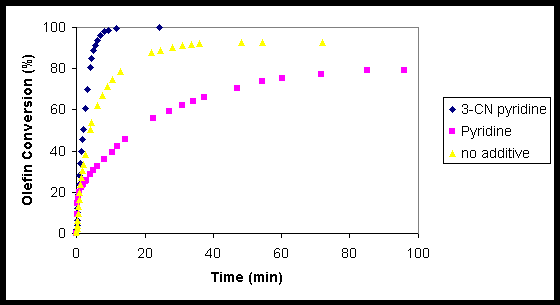
Figure 3. The epoxidation of 1-decene with 0.5 mol% MTO, 1.5 equiv 30% aq. H2O2, in CH2Cl2 (2M) is shown to proceed better with 10 mol% 3-cyanopyridine then without a pyridine additive. Surprisingly, the addition of 10 mol% pyridine was detrimental to he reaction and resulted in conversions worse then that of the blank reaction.
In the case of trans olefins, it was found that the 3-cyanopyridine was effective in the epoxidation of 3-trans-decene. An interesting effect was discovered when the amount of pyridine additive was varied. The addition of only 1 mol% 3-cyanopyridine allowed full conversion to the epoxide. In the original pyridine reaction, 1 mol% pyridine was insufficient for full conversion to the epoxide. The optimal amount of pyridine for full conversion of cis olefins is 12 mol%.
The pyridine additive was not the only variable studied. The MTO catalyst is expensive and an alternative rhenium catalyst was desired to reduce the cost of the catalyst. One of the main reasons many inorganic rhenium sources are inaccessible in our current conditions is the presence of water. An anhydrous hydrogen peroxide source was studied for this application, bistrimethylsilylperoxide (BTSP)6. A number of inorganic rhenium sources were used including Re2O7, ReO3(OH), and ReO3. Though cheaper and more readily available then MTO, they less stable especially in the presence of water. The results of this screen are presented in Table 1.
Table 1. Results of Epoxidation with BTSP and various rhenium sources.
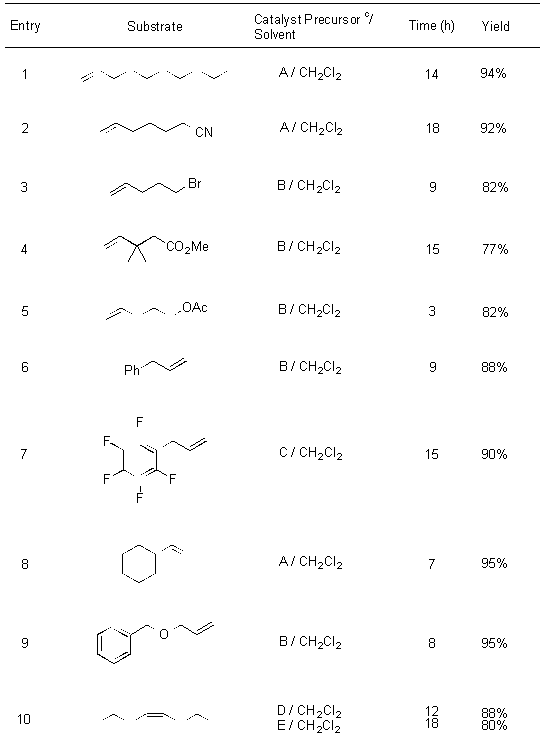

The use of automation in the optimization of reactions has been useful in accelerating the screening process. Robotics has been useful in the preparation of reactions, the unattended quenching of aliquots, and the preparation of GC samples. We have been able to consistently analyze 16 reactions and 192 timepoints a day. This translates to over 100 reactions and well over 1000 timepoints a week. With a number of robotic workstations that were not specifically designed to perform process research, we have been able to screen a large number of reactions and efficiently study an important catalytic transformation. One of the limitations of this process is the lengthy analysis time. The automated screening rate could be increased but the analysis would not keep stride. We have recently developed an analytical technique using flow-injection mass spectrometry to study the aminohydroxylation7. This allows the analysis time to be reduced to 2 minutes and greatly increases the potential throughput. In conjunction with an additive, mass spectrometry is capable of detecting epoxides through coordination-ionspray mass spectrometry8 and it may be possible to increase the rate of screening of epoxide formation with an accelerated analytical technique.
Acknowledgments: We thank the National Institutes of General Medical Sciences, National Institutes of Health (GM-28384), the National Science Foundation (CHE-9531152), the W. M. Keck Foundation, and the Skaggs Institute for Chemical Biology for providing financial support. JPC would like to thank the ARCS foundation for financial support and Eli Lilly for a graduate fellowship. We would like to thank Hans Adolfsson, Christophe Coperet, Joachim Rudolph, Laxma Reddy, and Andrei Yudin for their contributions to the development of the rhenium epoxidation system.
References
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with B0011 as the message subject of your e-mail.