[C0002]
Lixin Qiao,a* Rene Etcheberrigaray,a Alan P. Kozikowskib and Bryan Rothc
Received: 20 August 1999 / Uploaded: 24 August 1999
Research on cellular and molecular aspects of Alzheimer’s disease over the last two decades has resulted in a significantly improved understanding of this disease. Particular achievements have occurred in the areas of molecular genetics and the biology of amyloid. This research has strengthened the notion that A
b is a central player in the disease process.1-5 Despite the progress and the data accumulated, the understanding of the pathophysiological framework remains incipient. Consequently, rational drug design targeting specific pathophysiological events or chemical entities has seen little or slow progress. Additional difficulties arise from the fact that dynamic processes are difficult to assess in the AD brain as a number of signal transduction mechanisms are altered by postmortem events.6 In addition, AD brain tissue usually becomes available after a long period of the disease, at an advanced pathological state. Therefore, significant effort has been devoted to alternative approaches.6-10 A host of cellular and molecular changes ranging from nucleic acid defects to second messenger alterations have been described in peripheral cells of AD patients.6-8,10,11 Some alterations have also been observed in neuronal animal cells and brain.12-15 Of the many alterations, PKC seems uniquely situated in the cascade of events (normal and pathological) interacting with Ab, calcium homeostasis and ion channel function. Herein we propose a framework as an initial step for drug design targeting PKC.PROTEIN KINASE C
Protein kinase C is a ubiquitous family of enzymes involved in a number of cellular processes including (but not limited to) growth and differentiation, ion channel regulation, neuronal plasticity and memory storage.16-20 Some of the functions may be related to specific isozymes and /or their differential expression in different tissues and organs. PKC has also been the subject of attention in a wide range of pathological processes from cancer to nervous systems disorders.16, 20-22 A number of reports have identified alterations in protein kinase C in brains and fibroblasts of AD patients. Reduced levels of the particulate fraction in AD brains as measured by radioactive phorbol ester binding and in vitro phosphorylation of histone H I was originally reported by Cole et al.23 Phosphorylation of P86 was also found reduced in the cytosolic fraction.23 Additional reports have indicated that in particular the
bII isoform was significantly lower in the particulate fraction from AD hippocampal and cortical tissues.24 The opposite was true for the cytosolic fraction from cortex. The isoforms bI and a were also reduced in the particulate fraction from hippocampus of AD patients.24 Shimoama et al. confirmed a predominant involvement of the b isoform in AD brains.25 A more recent report found attenuated PKC activity and translocation in AD brains.26 However, the immunoreactivity (membranous fraction) of the a and g isozymes was found increased in the frontal cortex. This finding was attributed to reduced proteolytic activity and/or a compensatory response in AD.26 Some authors have suggested that PKC alterations may be an early event in AD.27 Levels of mRNA for the a isozyme were found somewhat reduced in AD brains, although there was enough overlap so the differences did not reach statistical significance.28 Other reports have not found salient differences in brain distribution of PKC in AD compared to controls.29,30 Studies in fibroblasts generally parallel those in brain tissues. Both immunoreactivity and protein phosphorylation were found reduced in familial and sporadic AD fibroblasts.31 More recently it was reported that the phorbol ester binding affinity and the phosphorylating activity (histone) were lower in cells from AD patients.32 Immunoblotting analyses revealed that the a isoform was primarily affected in AD fibroblasts.33,34PKC may also play a significant role in APP processing. APP is the precursor protein from which
b-amyloid originates and it possesses phosphorylation.1,35 PKC activators influence the type of secreted APP products. The APP ectodomain can be constitutively secreted by cleavage within the b-amyloid sequence (a-secretase) thus resulting in "non-amyloidogenic" processing. APP can also be internalized and degraded in an endosomal/lysosomal compartment generating fragments that contain the intact b-amyloid sequence, therefore resulting in potentially amyloidogenic (and possibly pathologic) processing.1,34,36 It has been shown that PKC activation can increase the rate of the non-amyloidogenic processing in cellular models. In addition, some studies have directly shown a reduction of the secretion of the b-amyloid peptide after phorbol treatment or activation of PKC.34,35,37,38 There are, however, two reports that show an increase in sAPP without changes in Ab.39,40 These apparently contradictory results could be due to tissue specific differences. In addition, Savage et al.41 reported a phorbol-induced decrease in Ab species without a noticeable increase in sAPP secretion in mouse brain. Therefore, the general finding of reduced PKC amounts and/or activity in AD (brain and fibroblasts) is consistent with the apparently normal regulatory role of PKC favoring non-amyloidogenic processing of APP. It is worth noting that some of the Italian cell lines that showed the K+ channel defect and enhanced IP3 mediated calcium releases were the same ones in which defective PKC activity and immunoreactivity was demonstrated.32-34,42,43 Interestingly, in these AD fibroblasts the reduced PKC activity has been correlated with a reduced ability to secrete soluble APP.34,43 We report herein our recently designed novel PKC activators that are capable of increasing soluble APP.DESIGN CONCEPT OF PYRROLIDONE ANALOGS
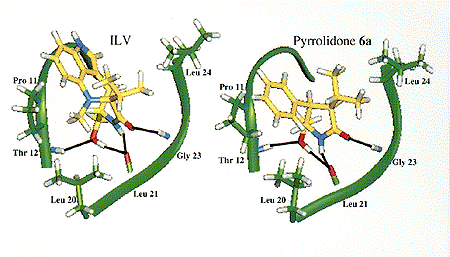
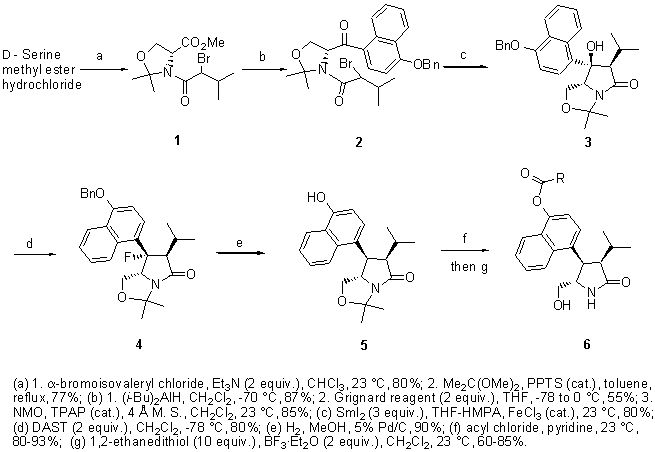
On the basis of the X-ray structure of PKC CRD2 (cysteine-rich domain) in complex with phorbol 13-acetate,44 we have determined how the high-affinity ligands ILV and the eight-membered ring benzolactam bind through a combination of molecular modeling and site-directed mutagenesis studies.45 To both simplify and to rigidify the ILV structure, conceptually we considered linking C-9 and N-13, as these atoms are close to one another in ILV's twist conformation (Figure 1a), to arrive at the pyrrolidone derivatives 6a (Figure 2). In order for this type of compound to interact efficiently with PKC, modeling studies revealed that the isopropyl and phenyl groups must be cis oriented and trans to the hydroxymethyl group. With this stereochemistry, the pyrrolidone is capable of engaging in the same hydrogen-bond network to PKC as identified for ILV (Figure 1). Its isopropyl group interacts with the side chain of Leu 24, thereby mimicking the isopropyl group of ILV. Also, its phenyl group is parallel to Pro 11, thus allowing for strong hydrophobic interactions. However, the important interaction of the N-methyl group of ILV with Pro 11 and Leu 20 is absent (the absence of this group in ILV results in a 100-fold reduction in potency46). In addition, the optimal water solubility values (log WS)47 can be adjusted by introduction of an appropriate substituent. This side chain generally enhances a ligand's binding affinity through interaction with the lipid membrane.
Figure 1. The overall features of the binding model for the ILV (left) and pyrrolidone derivative (right) in complex with PKC
d CRD2. 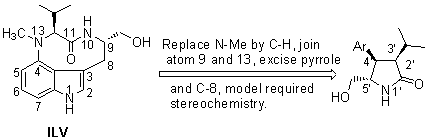
Based on our preliminary result of pyrrolidone 6a,48 molecular modeling
suggested that the replacement of the phenyl in 6a by an
LQ12, a substituted naphthyl pyrrolidone, was tested for its ability to displace phorbol 12, 13-dibutyrate (PDBU) binding from recombinant PKC
a. BL, an 8-membered benzolactam, was previously synthesized in our laboratory and shown to be a potent PKC activator. As is apparent from Table 1, LQ12 is a fairly potent PKC activator. It is about four-fold less active than BL.BL |
LQ12 |
|
Ki ± SEM (nM) |
23 ± 4 |
97 ± 35 |
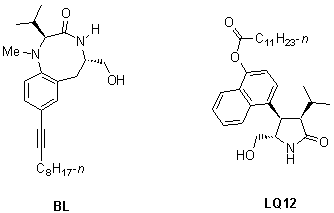
Materials and methods were used as published49 to evaluate sAPP secretion. Our previous results revealed that BL treatment induced elevations in sAPP among AD cell lines in a dose-dependent manner. Pretreatment with staurosporine, a potent PKC inhibitor, completely prevented the BL-induced sAPP secretion, supporting the involvement of PKC. As depicted in Figure 3,49 LQ12 significantly increased sAPP secretion in PC-12 cells and was found to be slightly less potent than BL. This effect on amyloid processing mirrors the binding results, further supporting the involvement of PKC.
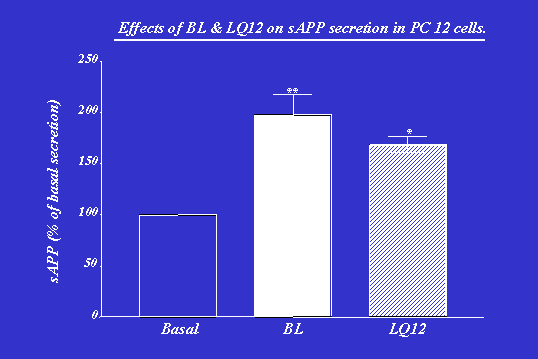
LQ12, a novel PKC activator, causes increased secretion of non-amyloidogenic sAPP in PC-12 cells. The elevated sAPP secretion may be also accompanied by a reduction of amyloidogenic fragments. The result further substantiates a key role for PKC in APP processing and, therefore, in AD pathophysiology. This study also suggests that PKC may be a useful target for preventing or slowing the pathophysiological process in AD. Furthermore, this novel compound offers the basis for drug design strategies targeted at PKC and APP processing that may significantly and beneficially alter the progression of this disease. Further studies aimed at optimizing the pyrrolidone structure so as to further maximize its activation of and therefore to maximize sAPP secretion are underway and will be released in due course.
1. Selkoe D.J. (1994) Normal and abnormal biology of the b-amyloid precursor protein. Annu. Rev. Neurosci. 17, 489-517.
2. Tanzi R. E., Kovacs D. M, Kim T., Moir R. D., Guenette S. Y., and Wasco W. (1996) The gene defects responsible for familial Alzheimer’s disease. Neurobiology of Disease 3, 159-168.
3. Selkoe D. J. (1997) Alzheimer’s disease: genotypes, phenotype and treatments. Science 275, 630-631.
4. Hardy J. (1997) Amyloid, the presenilins and Alzheimer’s disease. TINS 20, 154-159.
5. Lendon C. L., Ashall F., and Goate A. M. (1997) Exploring the etiology of Alzheimer disease using molecular genetics. JAMA 277, 825-831.
6. Gibson G., Martins R., Blass J., and Gandy S. (1996) Altered oxidation and signal transduction systems in fibroblasts from Alzheimer patients, in Life Sciences, vol. 59, Muller W. E., and Gispen W. H., eds., Elsevier, Heidelberg, Germany, pp. 477-489.
7. Baker A. C., Ko L-W., and Blass J. P. (1988) Systemic manifestations of Alzheimer's disease. Age 11, 60-65.
8. Scott R. B. (1993) Extraneuronal manifestations of Alzheimer's disease. JAGS 41, 268-276.
9. Huang H-M., Martins R., Gandy S., Etcheberrigaray R., Ito E., Alkon D. L., Blass J., and Gibson G. (1994) Use of cultured fibroblasts in elucidating the pathophysiology and diagnosis of Alzheimer's disease. Ann. N.Y. Acad. Sci. 747, 225-243.
10. Gasparini L., Racchi M., Binetti G., Trabucchi M., Solerte B., Alkon D. L., Etcheberrigaray R., Gibson G., Blass J., Paoletti R., and Govoni S. (1997) Peripheral markers in testing pathophysiological hypotheses and diagnosing Alzheimer’s disease. FASEB J. 12, 17-34.
11. Connolly G. P. (1998) Fibroblast models of neurological disorders: fluorescence measurement studies. TiPS 19, 171-177.
12. Good T. A., Smith D. O., and Murphy, R. M. (1996) b-amyloid peptide blocks the fast-inactivating K+ current in rat hippocampal neurons. Biophys. J. 70, 296-304.
13. Cole G., Dobkins K. R., Hansen L. A., Terry R. D., and Saitoh T. (1988) Decreased levels of protein kinase C in Alzheimer brain. Brain Res. 452,165-174.
14. Masliah E., Cole G., Shimohada S., Hansen L., DeTeresa R., Terry R. D., Saitoh T. (1990) Differential involvement of protein kinase C isozymes in Alzheimer’s disease. J. Neurosci. 10, 2113-2124 .
15. Shimohama S., Narita M. A., Matsushima H., Kimura J., Kamejama M., Hagiwara M., Hidaka H., and Taniguchi T. (1993) Assessment of protein kinase C isozymes by two -site enzyme immunoassay in human brains and changes in Alzheimer’s disease. Neurology 43, 1407-1413 .
16. Lee-Way J., and Saitoh T. (1995) Changes in protein kinases in brain aging and Alzheimer’s disease. Drugs and Aging 6, 136-149.
17. Olds J. L., and Alkon D. L. (1993) Protein kinase C: a nexus in the biochemical events that underlie associative learning. Acta Neurobiol. Exp. 53, 197-207.
18. Nishizuka Y. (1988) The molecular heterogeneity of protein kinase C and its implications in cellular regulation. Nature 334, 661-665.
19. Newton A.C. (1995) Protein kinase C: structure, function, and regulation. J. Biol. Chem. 270, 28495-28498.
20. Pascale A., Govoni S., and Battaini F. (1997) Age-related alterations of PKC, a key enzyme in memory processes: physiological and pathological examples. Molecular Neurobiol. In press.
21. O’Brian C.A., and Ward N. E. (1989) Biology of the protein kinase C family. Cancer and Metastasis Reviews 8, 199-214.
22. Nishino N., Kitamura N., Nakai T., Hashimoto T., and Tanaka C. (1989) Phorbol ester binding sites in human brain: characterization, regional distribution, age-correlation, and alterations in Parkinson’s disease. J. Molec. Neurosci. 1, 19-26.
23. Cole G., Dobkins K. R., Hansen L. A., Terry R. D., and Saitoh T. (1988) Decreased levels of protein kinase C in Alzheimer brain. Brain Res. 452,165-174.
24. Masliah E., Cole G., Shimohada S., Hansen L., DeTeresa R., Terry R. D., Saitoh T. (1990) Differential involvement of protein kinase C isozymes in Alzheimer’s disease. J. Neurosci. 10, 2113-2124 .
25. Shimohama S., Narita M. A., Matsushima H., Kimura J., Kamejama M., Hagiwara M., Hidaka H., and Taniguchi T. (1993) Assessment of protein kinase C isozymes by two -site enzyme immunoassay in human brains and changes in Alzheimer’s disease. Neurology 43, 1407-1413 .
26. Wang H-Y., Pisano M. R., and Friedman E. (1994) Attenuated protein kinase C activity and translocation in Alzheimer’s disease brain. Neurobiology of Aging 15, 293-298.
27. Masliah E., Cole G. M., Hansen L. A., Mallory M., Albright T., Terry R. D., and Saitoh T. (1991) Protein kinase C alteration is an early biochemical marker in Alzheimer’s disease. J. Neurosci. 11, 2759-2767.
28. Chachin M., Shimohama S., Kunugi Y., and Taniguchi T. (1996) Assessment of protein kinase C mRNA levels in Alzheimer’s disease brains. Jpn. J. Pharmacol. 71, 175-177.
29. Horsburg K., Deward D., Graham D., and McCulloch J. (1991) Autoradiographic imaging of [3 H] phorbol 12,13-dibutyrate binding to protein kinase C in Alzheimer’s disease. J. Neurochem. 56, 1121-1129.
30. Clark E. A., Leach K. L., Trojanowski J. Q., and Lee V. M-Y. (1991) Characterization and distribution of the three major human protein kinase C isozymes (PKCa, PKCb, PKCg) of the central nervous system in normal and Alzheimer’s disease brains. Lab. Invest. 64, 35-44.
31. Huynh T. V., Cole G., Katzman R., Huang K-P., and Saitoh T. (1989) Reduced protein kinase C immunoreactivity and altered protein phosphorylation in Alzheimer’s disease fibroblasts. Arch. Neurol. 46, 1195-1199.
32. Govoni S., Bergamaschi S., Racchi M., Battaini F., Binetti G., Bianchetti A., Trabucchi M. (1993) Cytosol protein kinase C downregulation in fibroblasts from Alzheimer's disease patients. Neurology 43, 258-2586.
33. Racchi M., Wetsel W. C., Trabucchi M., Govoni S., Battaini F., Binetti G., Bianchetti A., and Bergamaschi S. (1994) Reduced protein kinase C immunoreactivity in fibroblasts from patients with Alzheimer's disease. Neurology 44, A164.
34. Racchi M., Wetsel W. C., Trabucchi M., Govoni S., Battaini F., Binetti G., Bianchetti A., and Bergamaschi S. (1994) Reduced protein kinase C immunoreactivity in fibroblasts from patients with Alzheimer's disease. Neurology 44, A164.
35. Gandy S., and Greengard P. (1994) Processing of Alzheimer Ab-amyloid precursor protein: cell biology, regulation, and role in Alzheimer disease. Int. Rev. Neurobiol. 36, 29-50.
36. Bhagavan S., Ibarreta D., Ma D., Kozikowski A. P., and Etcheberrigaray R. (1998) Restoration of TEA-induced calcium responses in fibroblasts from Alzheimer’s disease patients by a PKC activator. Neurobiology of Disease 5, 177-187.
37. Desdouits F., Buxbaum J. D., Desdouits-Magnen J., Nairn A. C., and Greengard P. (1996) Amyloid b peptide formation in cell free preparations: Regulation by protein kinase C, calmodulin, and calcineurin. J. Biol. Chem. 271, 24670-24674.
38. Efhimiopoulos S., Punj S., Manopoulos V., Pangalos M., Wang G. P., Refolo L. M., and Robakis N. K. (1996) Intracellular cyclic AMP inhibits constitutive and phorbol ester-stimulated secretory cleavage of amyloid precursor protein. J. Neurochem. 67, 872-875.
39. Fuller S. J., Storey E., Li Q-X., Smith A. I., Beyreuther K., and Masters C. L. (1995) Intracellular Production of bA4 Amyloid of Alzheimer’s disease: modulation by phosphoramidon and lack of coupling to the secretion of the amyloid precursor protein. Biochemistry 34, 8091-8098.
40. LeBlanc A. C., Koutroumanis M., and Goodyer C. G. (1998) Protein kinase C activation increases release of secreted amyloid precursor protein without decreasing Ab production in human primary neuron cultures. Journal of Neuroscience 18, 29097-2913.
41. Savage M. J., Trusko S. P., Howland D. S., Pinsker L. P., Mistretta S., Reaume A. G., Grrenberg B. D., Siman R., and Scott R. W. (1998) Turnover of amyloid b-protein in mouse brain and acute reduction of its level by phorbol ester. The Journal of Neuroscience 18, 1743-1752.
42. Hirashima N., Etcheberrigaray R., Bergamashi S., Racchi M., Battaini F., Binetti G., Govoni S., and Alkon D. L. (1996) Calcium responses in human fibroblasts: a diagnostic molecular profile for Alzheimer’s disease. Neurobiology of Aging 17, 549-555.
43. Govoni S., Racchi M., Bergamaschi M., Trabucchi M., Battaini F., Bianchetti A. and Binetti G. (1996) Defective Protein Kinase C a leads to impaired secretion of soluble b-amyloid precursor protein from Alzheimer’s disease fibroblasts. Ann. N. Y. Acad. Sci. 777, 332-337.
44. Zhang, G.; Kazanietz, M. G.; Blumberg, P. M.; Hurley J. H. Cell 1995, 81, 917.
45. Kozikowski A. P., Wang S., Ma D., Yao J., Ahmad S., Glazer R. I., Bogi K., Acs P., Modarres S., Lewin N. E., and Blumberg P. M. (1997) Modeling, chemistry, and biology of the benzolactam analogues of ILV. 2. Identification of the binding site of the benzolactams in the CRD2 activator-binding domain of PKCd and discovery of an ILV analogue of improved isozyme selectivity. J. Med. Chemistry 40, 1316-1326.
46. Irie, I.; Okuno, S.; Kajiyama, S.; Koshimizu, K.; Nishino, H.; Iwashima, A. Carcinogenesis 1991, 12, 1883.
47. Marquez, V.; Lee, J.; Sharma, R.; Teng, K.; Wang, S.; Lewin, N. E.; Bahador, A.; Kazanietz, M. G.; Blumberg, P. M. Bioorg. Med. Chem. Lett. 1994, 4, 355.
48. Qiao, L.; Wang, S.; George, C.; Lewin, N. E.; Blumberg, P. M.; Kozikowski, A. P. J. Am. Chem. Soc. 1998, 120, 6629.
49. Ibarreta D., Duchen M., Ma D., Qiao L., Kozikowski A. P., and Etcheberrigaray R. (1999) Benzolactam (BL) enhances sAPP secretion in fibroblasts and PC12 cells. NeuroReport 10, 1035-1040.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with C0002 as the message subject of your e-mail.