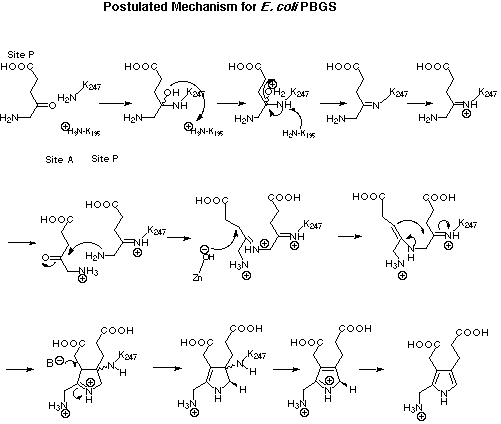
Figure 26: Proposed mechanism for the biosynthesis of porphobilinogen (4).[49]
Conclusions
Conclusions Based on our inhibition results and incorporating the information from the X-ray structures the following mechanism can be proposed for porphobilinogen synthase (see Figure 26).[49]
Figure 26: Proposed mechanism for the biosynthesis of porphobilinogen (4).[49]
The reaction sequence starts with the formation of the first Michaelis-Menten complex at the P-site of the enzyme. The first substrate forms then the Schiff base with the active-site lysine. As proposed by Jordan and Cooper the role of the second lysine could be to play the role of a proton donor and a proton acceptor during the formation of the Schiff base.[49] At this stage the second Michaelis-Menten complex is formed at the A-site. Once the second substrate has been recognised by the active site the crucial bond forming between the two substrate molecules has to occur. The Schiff base connecting the two 5-aminolevulinate molecules (5) should then be transformed into the corresponding enamine. The enamine contains all the reactive centres in the arrangement necessary to create the crucial carbon carbon bond. Once the carbon carbon bond is formed a series of reversible but chemically straight forward steps will lead to the product porphobilinogen (4). A plausible sequence could be: deprotonation a to the iminium ion forming the enamine, followed by elimination of the amino group of the active site lysine, finally the protonated form of porphobilinogen is deprotonated by a base at the active site of the enzyme, thereby forming porphobilinogen (4) bound to the enzyme.
This proposal incorporates a series of observations from the X-ray structures and is in accordance with most of our inhibition results. At the same time a series of new questions have arisen and a few chemical problems have still not found an answer so far.
The following structural and kinetic information has been incorporated:
The second lysine group found at the active site has a role as proton shuttle and as group influencing the pKa of the active site lysine. The attribution of this role is largely inferred indirectly. It is at the moment not possible to exclude that this lysine plays a different role e.g. forming a Schiff base with the second substrate. There is a major role attributed to the active site Zn. The active site Zn should be essentially the porter of a judicially placed a base, the hydroxide ion, which plays a crucial role in creating regioselectively the enamine needed for the ring closure. A sort of secondary role can be attributed to the Zn ion, which would be that of complexing and thereby fixing the carbonyl group of the A-site substrate. Up to this point the arguments in favour of this mechanism are based on the X-ray structures of the porphobilinogen synthase isolated from yeast and Escherichia coli and from the structures obtained by co-crystallisation with levulinic acid. The precise role of each unit mentioned in the mechanism above, is reasonable, for some of these roles even good analogies from other enzymes exist. The distances within the active cleft allow to propose these roles.
The contribution of the inhibition studies is mostly indirect, but rather compelling. In our trials to study analogues of the proposed intermediates we were confronted with the fact, that all the analogues to the Shemin sequence of events were weak inhibitors, whereas the inhibitors based on the Jordan sequence showed good inhibition potency or became even irreversible inhibitors. This analysis is the consequence of the study of more than 100 compounds and is therefore based on a reasonable large data set. Our results are compatible with the sequence of events first proposed by Jordan. The inhibition kinetics alone will however not be able to prove the sequence of events.
Problems arise from three lines of arguments: 1) What will be the mechanism of Pseudomonas aeruginosa? 2) What is the "chemical" logic of the proposed sequence? 3) What is the relation of the actual biochemical mechanism to the hypothesis of a prebiotic formation of tetrapyrroles?
The problem posed by the structure of Pseudomonas aeruginosa is quite evident. The position of the Mg2+ observed and thereby the role attributed to Mg-ion in porphobilinogen synthase from Pseudomonas aeruginosa is completely different from that of Zn2+ in the enzymes isolated from yeast and Escherichia coli. A probable consequence of this difference could be that the mechanisms are different for the Zn-enzymes and for the Mg-enzymes which would give an enhanced importance to the classification of porphobilinogen synthase according to their metal content made earlier by Jaffe.[41,56]
A question indirectly related to the first problem but already touching the problem of the "chemical" logic is the good recognition of the carbonyl group by the enzyme. All substrate analogues with good inhibition behaviour have to have the g-keto function. In the mechanistic proposal the recognition of the keto function is relatively vaguely defined. The Zn2+ is co-ordinated in a tetrahedral geometry to three cysteines and to a water molecule. Already Shoolingin-Jordan and Cooper observe that co-ordination only to cysteines is unusual for an active site Zn cation.[49] It is also not evident how the Zn2+ may play the double role of the porter of the hydroxide base, as well as the role of the co-ordination site. At a first glance it seems reasonable that the Zn cation plays either one of these two roles, but it is not at all obvious how the active site Zn2+ can play both roles at the same time?
The major problem in connection with the proposed sequence is the question of the ring closing process. This process does not follow Baldwins rules. It has been shown that this ring closure is possible but it is not a favourable process.[57] The obvious questions in this context are to find out how the enzyme has learned to overcome the unfavourable stereoelectronics of the ring closure and how the enzyme manages to accommodate the changes in hybridisation and therefore in the conformation of a relatively tightly bound intermediate. Or in other terms how can the enzyme keep the strong interaction with the two carboxylic acids during this process if the spatial arrangement of the central part of the molecule changes completely. A related question concerns the deprotonation which leads to the enamine. From the solution studies it is clear that deprotonation is thermodynamically favoured towards the ammonium group of 5-aminolevulinate.[58] The obvious answer to this question is that the enzyme has only a basic site juxtaposed for the deprotonation at carbon 3. Our actual knowledge does not allow us confirm this hypothesis, especially because we do not know how the keto function is interacting with the enzyme. So the conclusion from the mechanistic analysis of the proposed mechanism is that once the carbon carbon bond is formed the mechanism is chemically reasonable and easy to understand. The problems posed are why is the crucial step coming relatively late in the mechanism and how does the enzyme circumvent a series of stereoelectronic obstacles connected with this mechanism.
The final question arising with this mechanism connects today's enzymatic mechanisms with the postulated prebiotic formation of tetrapyrroles.[23,59] Assuming that the hypothesis of the prebiotic formation of tetrapyrroles is correct, then there are immediately two questions which are connected with the enzymatic formation of porphobilinogen (4): as the chemical reactivity of 5-aminolevulinate (5) does not give any hint towards a pre biotic formation of porphobilinogen (4).[60] the question is: does the biochemical mechanism give us any hint towards the prebiotic formation of porphobilinogen (4) or an adequate prebiotic analogue?[59] At this stage the answer to this question is probably no. Then the second question is obvious: how did nature make the switch from prebiotic conditions to an enzyme controlled synthesis? The studies of the structure of the enzyme have not created a clear cut answer. At the moment we can only speculate and try to undertake experiments supporting our speculations. But the gap between our knowledge about prebiotic processes and the knowledge accumulated about the biochemistry seems to increase. This gap is all the more embarrassing as the synthesis of porphobilinogen (4) is central to all tetrapyrrole synthesis. We have good evidence that starting from porphobilinogen (4) tetrapyrrole synthesis is in principle easy to explain be it prebiotic, be it chemical, be it biochemical. It would therefor be important to be able to show that also the synthesis of porphobilinogen (4) itself is easy.
In conclusion the sum of chemical, biochemical and in the last two years X-ray studies
has increased our knowledge about porphobilinogen synthase considerably. We are not yet in
the position to have firm proof of one mechanism, but at least for porphobilinogen
synthase from Escherichia coli the combination of structural data with the results of
inhibition studies allows to propose the Jordan sequence as the most probable. The tools
are probably now available to solve the question of the sequence of the enzyme catalysed
reaction, by trying to co-crystallize the enzyme with some of the analogues of the
postulated intermediates. We will hopefully be able to obtain definitive proof soon. We
will then be able to infer reasonable answers to some of the questions posed above.
The author is grateful to the Swiss National Science Foundation, the Novartis Agro, the
KGF and the Hoffmann-La Roche company for generous support of this work and to my
dedicated co-workers: Anne Meunier, Rainer Lüönd, Hugo Bertschy, Matthias Henz, Thomas
Engeloch, Pavel Bobal, Janette Bobalova, Caroline Jarret, Frédéric Stauffer, Eleonora
Zizzari for their excellent efforts.
![]() "A Novel Synthesis of Porphobilinogen: Synthetic And Biosynthetic
Studies"
"A Novel Synthesis of Porphobilinogen: Synthetic And Biosynthetic
Studies"
Christiane Bobillier Neier / August 1999