Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0012]
Stereoinduction in the Tosyl Radical-Mediated Cyclization
of Polysubstituted 1,6-Dienes Derived from
D-Glucose and D-Mannose
Dora-Marina Gutierrez-Avella, Michele Bertrand*, Robert Nouguier*
Laboratoire de Chimie Moleculaire Organique, UMR 6517, Boite 562, Faculte des Sciences St-Jerome, Universite d�Aix-Marseille III, Avenue Normandie-Niemen, 13397 Marseille, Cedex 20, France.
Fax: (33) 4 67 09 44, E-mail : [email protected]
Received: 12 July 2000 / Uploaded: 29 July 2000
INTRODUCTION
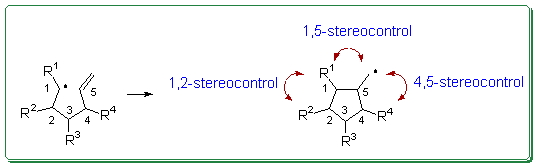
Radical cyclizations offer an easy access to five- and six-membered rings, and their applications to the synthesis of complex molecules are now incredibly numerous.1 The control of stereochemistry in theses processes is generally high and models have been designed which allow to make fairly good predictions.2-3 Carbohydrates have been used as chiral template for the construction of highly substituted cyclopentane derivatives and they provide a variety of stereochemically well defined starting materials which could be used to evaluate the influence of polysubstituted ethylenic tethers on the stereochemical balance of 5-exo ring closures.4
The fate of 5-hexenyl radicals leading to disubstituted cyclopentanes is well established,2,3 and can generally be accounted for through the determination of the contribution of the various chair-like and boat-like transition states at a given temperature. As an example, 5-hexenyl radicals bearing only one substituent in position 1 lead to cis cyclopentanes (Scheme 1, R2 = R3 = R4 = H). As far as polysubstituted systems are concerned, theoretical calculations are not very easily carried out, there is still a need for qualitative predictive rules. For instance, with secondary radicals, it can be inferred from literature data that 1,2-stereocontrol is exclusively trans, while at the same time 1,5-stereocontrol is only predominantly cis (cis:trans � 70:30) (Scheme 1, R3 = R4 = H)5-6 A similar trend is observed with 4,5-stereocontrol which is generally exclusively trans while simultaneously the relative configuration of the substituents in position 1 and 5 is only partially controlled (Scheme 1, R2 = R3 = H).7
Scheme 1
In an elegant study, performed in parallel on cyclic radicals derived from glucose, mannose and galactose respectively, Rajan-Babu has demonstrated that A1,3-strain can force the cyclization to proceed through a boat-like transition state, which therefore gives rise exclusively to a 1,5-trans product (this is observed in the glucose series).8 In a related study, we have shown that 1,2-stereocontrol could also exert a constraint on the 5-exo ring closure of a secondary radical onto a cyclohexene, sufficient to induce a 1,5-trans relationship between the substituents in the major product.6a
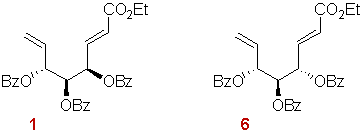
Figure 1
As a continuation of our studies on the cyclofunctionalization of 1,6-dienes,6,9 we have investigated the behaviour of substrates 1 and 6 (Figure 1), prepared respectively from D-mannose and D-glucose, with respect to the addition of TsSePh. These reactions lead to potentially useful polyfunctionalized cyclopentanic synthons, but the practical value of this methodology relies on the degree of stereoselectivity in the cyclization step. Contrary to the system developed by Rajan-Babu where the secondary radical is cyclic,8 in this case, the radical formed through the initial addition of tosyl radical possesses an entirely flexible ethylenic tether. These substrates were expected to lead to comparative information about the weight of the different types of interactions in the transition structures.
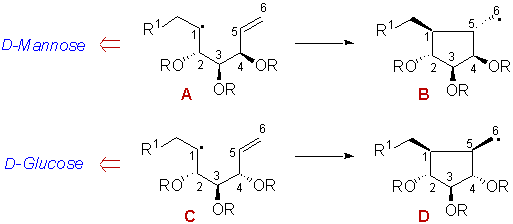
Scheme 2
On the ground of the above mentioned body of data, one can conclude that both 1,2- and 4,5-stereocontrols should exert a major influence on the stereochemical balance. As exemplified in Scheme 2, one would expect any glucose derived radical of type C to lead to D as the major isomer of the cyclized radical. This would result from the synergy of 1,2-, 4,5- and 1,5-stereocontrols. Indeed there are already data in the literature which confirm the prediction,7c, 8, 10-13 and according to which, D would account for approximately 70% of the isomeric mixture.
In the case of a radical of type A, one would expect the major product to result from radical B. Although it is an oversimplified view of the cyclization, this would be observed, only if 1,2- and 4,5-stereocontrols would overcome together the preference for forming products with a 1,5-cis stereochemistry, since there is no cooperative effect in this particular case. In the mannose series, there are no related examples in the literature with a C=C bond as the radical acceptor. The only related data, referring to cyclizations onto a C=C bond, that are available in the literature concern SmI2-mediated 5-exo cyclizations of sugar derived ethylenic aldehydes.14 Owing to the ability of samarium to form chelates with the basic oxygen atoms, the stereochemical feature of theses reactions cannot be compared to any of the previous ones.
RESULTS
Dienes 1 and 6 were prepared from methyl 2,3,4-tri-O-benzoyl-6-deoxy-6-iodo-a-D-manno- and glucopyranoside respectively, through a zinc-mediated fragmentation15 followed by a Horner-Emmons reaction with triethyl phosphonoacetate. The choice of TsSePh, rather than a tosyl halide, as the precursor of tosyl radical is based on the greater stability of the resulting selenides with respect to elimination compared to the halides. The cyclization resulted in the creation of three new stereogenic centers (Scheme 3), the chiral center a to the ester group was subsequently removed through the reduction of the phenylselenide with tributyltin hydride. In order not to perturb the diasteromeric ratio, the reduction was performed immediatly after the addition TsSePh was completed, by adding Bu3SnH in excess and a catalytic amount of AIBN, to the mixture of isomeric adducts in benzene at reflux.

Scheme 3
Cyclofunctionalization of 1 (R1=OBz, R2=H) and 6 (R1=H, R2=OBz)
In both cases the reaction led to a mixture of four stereoisomers. The distribution of the product is given in Schemes 4 and 5 together with relevant 13C NMR data.
Diene 1 led to compounds 2-5 in a 25:12:38:25 ratio with a 80% overall yield.

Scheme 4
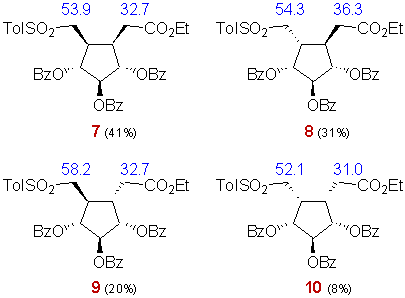
Diene 6 led to compounds 7-10 in a 41:31:20:8 ratio with a 64% overall yield.
Scheme 5
The structures were assigned from NMR data on the ground of the shielding of the carbon atoms of the two methylene groups. The carbon atom bearing the tosyl group, designed as C7, is deshielded in compound 4 (59.2 ppm) since it does not experiment any g-gauche effect. On the contrary it is particularly shielded due to the presence of two vicinal substituents in a cis position in compound 5 (52.2 ppm). For the same reasons, the carbon atom bearing the ethoxycarbonyl group (C6) is shielded in compound 2 (30.8 ppm) whereas it is deshielded in compound 4 (36.1 ppm). The chemical shifts of C7 observed for 2 and 3 are nearly identical (55.9 and 55.0 ppm respectively), the same observation applies to the chemical shifts of C6 observed for 3 and 5 (33.8 and 33.9 ppm respectively).
Structures 7-10 were assigned similarly, on the ground of the chemical shifts of C6 and C7. C7 is particularly deshielded in 9 (58.2 ppm) due to the absence of any vicinal cis substituent, so is the case for C6 in compound 8 (36.3 ppm). Both C7 and C6 are particularly shielded in 10 (52.1 and 31.0 ppm respectively) where each one has two cis neighbouring groups. C6 has the same chemical shift in 7 and 9 (32.7 ppm) since it is influenced by only one cis neighbouring group in both isomers. The same is true for C7 in compounds 7 and 8.
DISCUSSION
Qualitatively the predictions have been verified for both substrates, as far as the major component is concerned. But in both cases the major isomer accounts for approximately 40% of the mixture which indicates a rather low selectivity. It must be pointed out that in the case of 6, the diastereomeric distribution is somewhat different from those given in the literature for related examples,8, 10 in particular, the fourth isomer (that is the one corresponding to 10), has been reported only for reactions involving 3-deoxygenated radicals.11-12 This might be ascribed to the nature of the hydroxyl protective groups, but we believe that it is rather related to the nature of the substituent in position 1.
In fact each isomer can result from two different routes, the first one going through a chair-like transition state and the second through a boat-like transition structure. The eight possible transition structures are represented in Scheme 6.
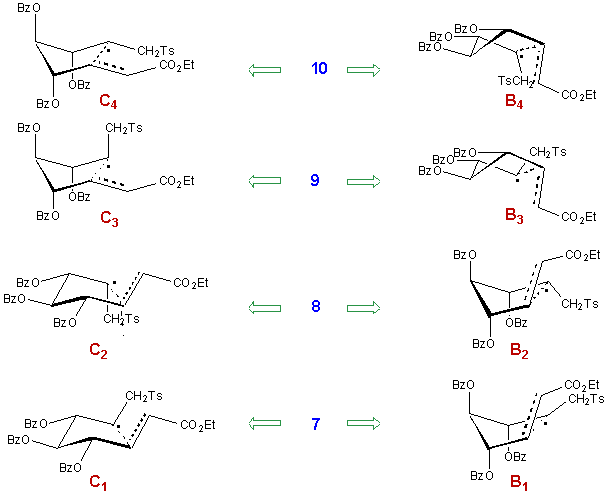
Scheme 6
The first statement is that in the glucose series, taking into account only the chair-like transition states would lead to a correct prediction of the product distribution. The most stable transition structure C1, which has no axial susbtituent, accounts for the preferential formation of 7. Going from zero to three axial substituents, destabilizes the chair-like transition structure as if one axial substituent would cost approximately 11% in term of contribution. It can be noted that conformers C1 and C2 correspond to the mimimal A1,3-strain for the allylic ether moiety (Figure 2).16 On the opposite, conformers C3 and C4 are likely to be very energetic.

Figure 2 : Preferred Conformation of Allylic Ethers
The fact that the predictions based strictly upon steric interactions in the chair-like transition states are correct is probably related to the destabilization of all boat-like structures owing to their energetic conformation around the C4-C5 bond.
Owing to the pseudo-equatorial position of the substituent in position 3 in the lowest energy conformers of the transition state, it is likely that the corresponding deoxygenated substrate would lead to a similar distribution of isomers. This has been verified by the studies directed towards the synthesis of prostacyclins that were conducted by Rokach11 and by Rossi.12 Provided the remaining hydroxyl groups are protected, the modification of the diastereomeric composition is only slightly affected by the nature of the protective groups, the choice of which is mainly governed by how easily they can be introduced or removed.
The analysis is more complex in the case of diene 1. The eight transition structures are shown in Scheme 8.
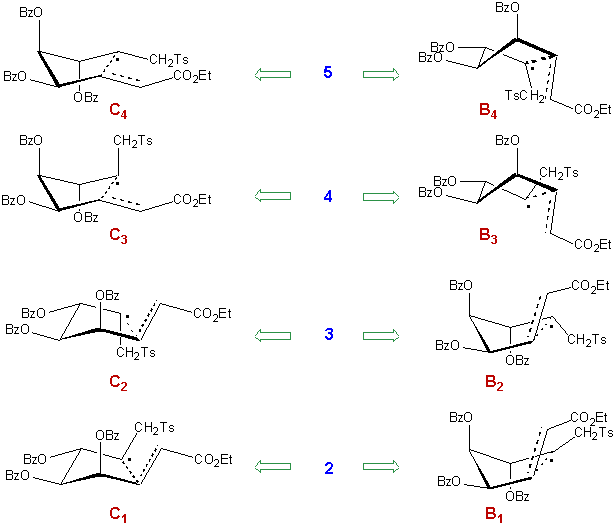
Scheme 8
In agreement with the above discussion, the contribution of 2 and 3 should be low. And one could without risk predict that 3 should be the minor product due to the unfavourable conformation of the allylic ether moiety and to the presence of two axial substituents both in C2 and B2.
There are reached the limits of qualitative predictions. It is really difficult to determine which of the remaining possible conformations of the transition structure is the most stable. There is no obvious reason why 5 should be the major component of the isomeric mixture. It seems that in this case, the presence or the absence of a substituent at C3 should influence the ratio of the various isomers more strongly than in the glucose series, since it occupies an axial position in the chair-transition structures contributing to the formation of the major products. But this remains to be investigated.
CONCLUSION
The cyclization of sugar derived flexible 1,6-dienes, mediated through the addition of sulfonyl radical presursors, allows in one step the preparation of cyclopentitols bearing substituents that open further transformations inherent to the introduction of a sulfone group. However, it is still difficult to make good predictions as regard to the diastereoselectivity of these processes. The best stereoinduction can be expected from the glucose series. It is to be noted nevertheless that, the simple statement that 1,2-trans and 4,5-trans stereocontrols have a predominant effect, allows to predict with a fairly good certitude the structure of the major isomer.
REFERENCES
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0012 as the message subject of your e-mail.