[A0019]
The 1,2,4-Triazolyl Cation: Thermolytic and Photolytic Studies
Rudolph A. Abramovitch1*, Harry H. Gibson, Jr.2 and Santiago Olivella3
1Department of Chemistry, Clemson
University, Clemson, SC 29634-0973
2Department of Chemistry, Austin College, Sherman, TX 75090-4440
3Centre de Recerca en Química Teòrica, Departaments de Quimica Física and
Quimica Organica, Universitat de Barcelona, Marti i Franques 1, 08028-Barcelona,
Catalonia, Spain
Received: 2 August 2000 / Uploaded: 3 August 2000
Abstract: The generation of the 1,2,4-triazolyl cation (1) has been attempted by the thermolysis and photolysis of 1-(1,2,4-triazol-4-yl)-2,4,6-trimethylpyridinium tetrafluoroborate (2) and the thermolysis of 1- and 4-diazonium-1,2,4-triazoles, using mainly mesitylene as the trapping agent. Thermolysis of (2) gave mostly 1,2,4-triazole, together with 3-(1,2,4-triazol-4-yl)-2,4,6-trimethylpyridine, 4-(1,2,4-triazol-4-ylmethyl)-2,6-dimethylpyridine and 4-(2,4,6-trimethylbenzyl)-2,6-dimethylpyridine. Thermolysis of each of the diazonium salts in mesitylene again gave mainly triazole together with very low yields of 1-(1,2,4-1-yl)-2,4,6-trimethylbenzene and the corresponding -4-yl isomer in about the same ratio. On the other hand, photolysis of (2) in mesitylene gave mainly 1-(1,2,4-triazol-1-yl)-2,4,6-trimethylbenzene. A photoinduced electron transfer from mesitylene to (2) has been observed and preliminary laser flash photolyses of (2) and the corresponding 2,4,6-triphenylpyridinium salt have been carried out. The observed transients are explained as arising from the first excited states of the pyridinium salts rather than from (1). Ab initio MO calculations are reported and indicate that the predicted electronic ground-state of the triazolyl cation is a triplet state of B1 symmetry with five p electrons, which corresponds to a diradical cation (1c). Possible mechanisms for the formation of the various products are proposed.
Introduction
Nitrenium ions have received much attention in recent years:1 synthesis and synthetic applications,2 and their implication as 'ultimate carcinogens' from aromatic amines.3 Alkyl-,2e aryl-,1,2a,c acyl-,4 and methoxynitrenium ions 5 have all been generated, but there was no reference to the production of 5-membered heteroaromatic s-nitrenium ions until 1991, when we published a preliminary communication on the subject.6a The only relevant papers published until then dealt with the calculations of the orbital energy levels of the p-cations of the 1H-imidazol-1-ylium, -pyrazolylium and -pyrrolylium,7 and the generation of such p-cations stabilized by two or more electron-donor dimethylamino groups.8
We initiated a study of the possible thermolytic generation and reaction of the 4-(1,2,4-triazolyl) cation (1) from readily available and inexpensive 4-amino-1,2,4-triazole,6, 9 and have extended it to the photolysis of suitable nitrenium ion precursors. We now describe these studies in full.
The original conception was that should a nitrenium ion (1a) be formed by cleavage of a s-bond to N4, it could possibly undergo electronic reorganization to the 5p diradical cation (1b), to an isomeric 5p diradical cation (1c), to the 4p cation (1d) (antiaromatic if fully conjugated), and to the s-cation (1e), isomeric with (1a). In addition, singlet and triplet multiplicities would be possible.10
We have shown that N-amino- and N-acylaminopyridiniums produce nitrenium ions on thermolysis. Thus, heating 1-(N-phthalimido)-2,4,6-triphenylpyridinium tetrafluoroborate in mesitylene/(CF3)2CHOH at 180�C gave N-mesitylphthalimide, while thermolysis or photolysis in anisole gave phthalimidoanisoles in 71.6% yield.4b Photolysis of 1-(N-phthalimido)-2,4,6-trimethylpyridinium tetrafluoroborate in anisole containing (CF3)2CHOH and CF3CO2H gave, following hydrazinolysis, the anisidines (94.5%) (o-: m-: p- :: 66.4: 8.3: 20.3), clearly suggesting that an electrophilic reactive intermediate (the diacylnitrenium ion) was generated.
Results
The 1-(1,2,4-triazol-4-yl)-2,4,6-trimethylpyridinium tetrafluoroborate (2) was readily prepared in 68% yield from 2,4,6-trimethylpyrilium tetrafluoroborate and 4-amino-1,2,4-triazole (3). Thermolysis of (2) under N2 in degassed mesitylene (containing sufficient hexafluoroisopropanol to bring (2) into solution at room temperature) at 208�C for 7 h gave a mixture of at least 8 compounds, from which could be isolated 3-(1,2,4-triazol-4-yl)-2,4,6-trimethylpyridine (4) (3.9%), 4-(1,2,4-triazol-4-ylmethyl)-2,6-dimethylpyridine (5) (4.7%), 4-(2,4,6-trimethylbenzyl)-2,6-dimethylpyridine (6) (20.9%), and 1,2,4-triazole (7) (60.5%). No 2-(1,2,4-triazol-4-yl)mesitylene (8) was detected. An authentic sample of the latter was synthesized (44% yield) from N,N-dimethylformamide azine (9) and 2-aminomesitylene in the presence of p-toluenesulfonic acid.6b

The structure of (4) was established unambiguously by single crystal X-ray diffraction (Fig. 1).

Fig. 1. Thermal ellipsoid plot (50% probability) of 4 showing the atom numbering scheme.
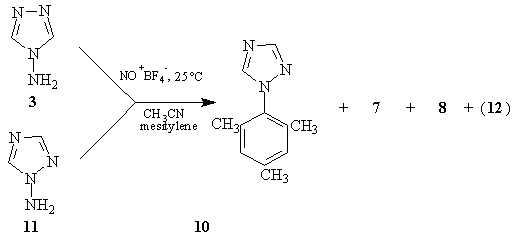
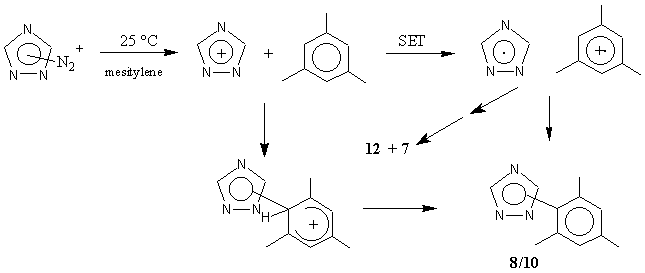
An alternate route to the 4-(1,2,4-triazoyl) cation was explored, namely the diazotization of 4-aminotriazole.9 Treatment of (3) with nitrosonium tetrafluoroborate in dry acetonitrile gave only (7), and no Ritter reaction-type products were detected. When the same reaction was carried out in the presence of mesitylene at room temperature, (7) was again isolated (77%) and nitrogen and nitrous oxide (the latter detected by infrared spectroscopy) were evolved. Remarkably, 1-(1,2,4-triazol-1-yl)-2,4,6-trimethylbenzene (10) was also isolated (1%), as were traces of 1-(1,2,4-triazol-4-yl)-2,4,6-trimethylbenzene (8) and the unsymmetrical mesitylene dimer (12). An authentic sample of (10) was synthesized from 2,4,6-trimethylphenylhydrazine hydrochloride and s-triazine using the general procedure of Grundmann and Ratz.11 The same products, in approximately the same ratio, were obtained when (3) was treated with isoamyl nitrite in the presence of acetic acid and excess mesitylene, or when 1-amino-1,2,4-triazole (11) was treated as above with NO+BF4-. No (10) was detected in the thermolysis of (2) in mesitylene.
We now turned to an exploration of the photolytic decomposition of the N-triazolopyridinium salts. Not unexpectedly, the results are quite different from the thermolytic ones. Thus, when (2) in acetonitrile and mesitylene was photolyzed (300 nm) for 30 h, the major products observed were (10) (48%), (7) (18%), and 2,4,6-collidine (81%). Trace amounts of (8) (the isomer of (10)) were detected by GC/MS. Under some conditions, small amounts of the mesitylene dimer (12) (comparison with know authentic sample),4a, 11 as well as the collidine-derived products (4) and its isomer (13) were also observed by GC/MS.

Mesityltriazole (10) is also produced (15-20%), albeit less cleanly (the GC/MS spectrum is much more complex), by the photolysis of 1-(1,2,4-triazol-4-yl)-2,4,6-triphenylpyridinium tetrafluoroborate under the same conditions as were used for (2). The more complex reaction mixture may be partly owing to intamolecular attack on the 2-phenyl group, but other processes may well be involved. It has been observed6a that in the thermolysis of 1-(N-phthalimido)-2,4,6-triphenylpyridinium tetrafluoroborate (14) in toluene/(CF3)2CHOH, some 2-(o-aminophenyl)-4,6-diphenylpyridine was formed, as was a small amount of a mixture of o- and p-methyldiphenylmethane (cf. (12) above). The same reaction, but carried out in mesitylene in the presence of CF3CO2H gave, among other products, 2,4-diphenyl-6-(o-phthalimidophenyl)pyridine (8.5%) (15) and the corresponding para derivative (16) (trace).4b Photolysis of (14) in mesitylene /CF3CO2H gave (15) (25.7%) and (16) (12.6%) among others.4b

The formation of the 1-yl isomer (10) in reasonable amounts in the photolysis of (2) is noteworthy, compared with the formation of the 4-yl products (4) and (5) in the thermolysis (no (10) detected). This suggested the possibility that, in the photolysis, a concerted heterolysis of the pyridinium salt was assisted by the nucleophile mesitylene attacking the triazole at N-1. To test this possibility, an isomer of (2), 1-(1,2,4-triazol-1-yl)-2,4,6-trimethylpyridinium tetrafluoroborate (17) was synthesized from (11) and 2,4,6-trimethylpyrilium tetrafluoroborate. Its photolysis in mesitylene/actonitrile gave (10) (45 %) and traces of (8) and (12) (detected by GC/MS), in addition to (7) (19 %), thus negating this possibility.
The photolysis was carried out using a variety of solvent combinations (acetonitrile, hexafluoroisopropanol, mesitylene) and reaction conditions (degassed, in the presence of air, at various temperatures) and using either 254, 300, or 350 nm lamp. The highest yield of (10) (48%) was obtained from (2), using mesitylene in anhydrous degassed acetonitrile at 0 - 5 �C and the 300 nm lamp.
When (2) was photolyzed in pure acetonitrile, the major products were (7) (6%), 2,4,6-collidine (12%), and (13) (20%), with traces of (4) (GC/MS) (Table 1). To determine whether the formation of (13) involved an intra- or intermolecular process, the triphenylpyridinium salt was photolyzed in acetonitrile containing 2,4,6-collidine in excess and gave mainly (13). The corresponding photolysis of (2) in acetonitrile/2,6-lutidine gave a mixture of two isomers of (18), in addition to some (7) and (13).Thus, the formation of (13) is clearly an intermolecular process in the case of the above photolysis of the triphenylpyridinium, as is the formation of (18) from (2), suggesting that the formation of (13) from the trimethylpyridinium (2) in mesitylene is also an intermolecular process. The 4-yl isomers (4) and (8) are not photoisomerized to the corresponding 1-yl compounds (13) and (10), though (4) and (8) are slightly decomposed after irradiation with 300 nm light in degassed acetonitrile for 20h.
We attempted to trap cation (1) with a variety of nucleophiles. Photolysis of (2) in methanol-d4/acetonitrile (10:90 v/v) gave no methoxytriazole, but the yield of triazole itself increased from 6% (acetonitrile) to 24% (methanol-d4/acetonitrile). Photolysis of (2) in the presence of
a-methylstyrene, 1-trimethylsilyloxycyclohexene, 1-methoxy-1-trimethylsilyloxy-2-methylpropene,13a and vinyltrimethylsilane gave rather complex reaction mixtures containing triazole but none of the expected addition or substitution products.13b On the other hand, photolysis of (2) in the presence of allyltrimethylsilane (19) gave a very low yield (<<5%) of 1-allyl-1,2,4-triazole (20).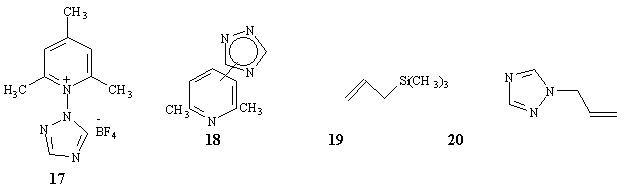
Table 1. % Yields of Products Formed from the Photolysis of (2) in Various Solvents.a
| Solvent | 7 | 2,4,6-Collidine | 13 | 12 |
| CD3CN | 6 | 12 | 20b | _ |
| CD3CN/MESc | 18 | 81 | traced | 48e |
| CD3CN/CD3ODf | 24 | 38 | 9.1 | _ |
| CD3OD | 42 | 38 | traced | _ |
a
Using 300 nm lamps in a Rayonet reactor at 5 �C. % Yields based on amount of decomposed pyridinium salt (in almost all cases >90%). bTogether with a trace of its isomer, (8). cMES = mesitylene. dLess than 1%; undetectable by NMR, observed by GC/MS. eTogether with a trace of (10). f90:10 (v/v).Allyltrimethylsilane and related silyl alkenes are quite reactive towards electrophiles, including carbocations, giving substitution products such as (20) with the loss of the trisubstituted silane.14 Allyltrimethylsilane also undergoes electron-transfer-initiated photoaddition to iminium salts via radical cation intermediates.15 The half-life of (2) in the presence of (18) depends on the concentration of the latter, which is compatible with either an SET process or with an assisted SN2' type reaction.16
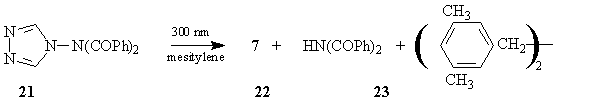
We also carried out experiments to see whether or not a 1,2,4-triazolyl free radical would react with mesitylene or with 2,4,6-collidine to form (8), (10), (4), or (16). Photolysis of (21) in mesitylene using 300 nm light17 proved to be a clean source of the desired radical, producing the products expected for such a radical: 1,2,4-triazole (7), dibenzoylimide (22), and the symmetrical 3,5-dimethylbenzyl radical coupling product (23). No (10) was detected.18
Results of Theoretical Calculations
Selected geometrical parameters (bond lengths in Å and bond angles in degrees) of the CASSCF(10,8)/6-31G(d)-optimized structures for the most relevant low-lying electronic states of (1) are shown in Figure 2. The harmonic vibrational analysis proved each of these structures to be a true potential energy minimum.19 Simple qualitative pictorial descriptions of the four highest occupied MOs of the calculated electronic states are shown in Figure 3. It is worth noting that the 6b2 MO is an out-phase combination of the nonbonding
s orbitals associated to the N1 and N2 atoms. The total and relative energies calculated at various levels of theory are shown in Table 2, along with the computed ZPVE.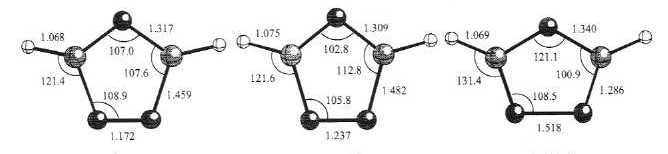
Fig. 2. Selected geometrical parameters of the CASSCF(10,8)/6-31G(d) optimized structures for the most relevant low-lying electronic states of the 1,2,4-triazolyl cation. Distances are given in angstroms and angles in degrees.
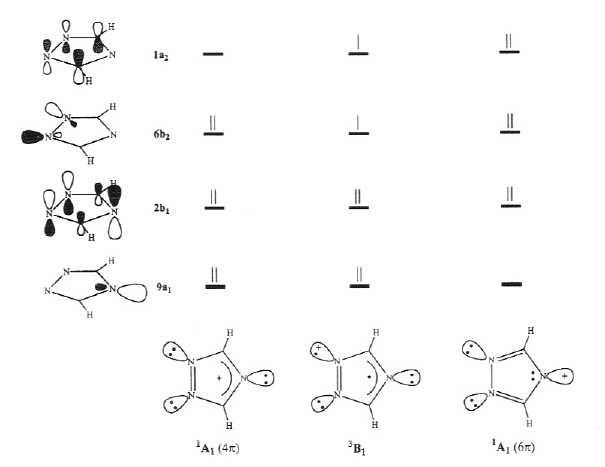
Fig. 3. Simple qualitative pictorial descriptions of the four highest occupied MOs of the most relevant low-lying electronic states of the 1,2,4-triazolyl cation.
Both the CASSCF(10,8) and CASPT2-g1 methods predict the electronic ground-state to be a triplet state of B1 symmetry with five
p electrons, which corresponds to diradical cation (1c), the lowest energy electronic singlet state being a closed-shell singlet of A1 symmetry with four p electrons, denoted 1A1(4p), which corresponds to p-cation (1d). Furthermore, the two methods agree in predicting the six p electron singlet state corresponding to s-cation (1a), denoted 1A1(6p) to lie high in energy above the ground state. On the basis of the calculated bond lengths, the electronic structure of 3B1, 1A1(4p), 1A1(6p) can be qualitatively represented by the structures shown at the bottom of Figure 3. The p electron system of the 3B1 and 1A1(4p) states can be formally regarded as formed by two separated (nonconjugated) moieties, namely, an allylic C3N4C5 unit with either three (3B1) or two (1A1(4p)) p electrons and a localized N1=N2 double bond. In contrast, the p electron system of the 1A1(6p) state can be described as two localized (nonconjugated) C=N double bonds plus a p nonbonding lone pair localized on N4, rather than as an aromatic 6p electron system.Table 2. Total Energies (E, Hartrees) Calculated at Different Levels of Theory and Zero-Point Vibrational Energies (ZPVE, kcal/mol) for Low-Lying Electronic States of 4-(1,2,4-Triazolyl) Cation
Ea |
||||
CASSCF(10,8) |
CASSCF(10,8)b |
CASPT2b |
||
| state | 6-31G(d) | cc-pVTZ | cc-pVTZ | ZPVEc |
| 3B1 | -239.88678 (0.0) | -239.97255 (0.0) | -240.75356 (0.0) | 28.2 |
| 1A1(4p) | -239.87949 (4.6) | -239.96756 (3.1) | -240.73749 (10.1) | 29.3 |
| 1A1(6p) | -239.84228 (27.9) | -239.92813 (27.9) | -240.71352 (25.2) | 29.8 |
aRelative energies in kcal/mol are shown in parentheses; bCalculated at the CASSCF(10,8)/6-31G(d) optimized geometries; cCalculated from the unscaled CASSCF(10,8)/6-31G(d) harmonic vibrational frequencies.
General Discussion
Thermolyses.
The most obvious difference between the thermolyses and photolyses of the N-triazolopyridiniums is that the triazole moiety does not lead to a triazolomesitylene in the former, but does in the latter. The absence of (8) in the thermolysis mandates the absence of cation (1) as a free entity under these conditions. The most likely route to (5) would go via the anhydro-base (24): a 1,5-sigmatropic shift (thermally allowed) would lead to (5). A similar 1,5-shift (also thermally allowed since the positive charge is not involved in the reorganization of the electrons) would lead to (4). We suggest that (6) may be formed from (24) by an attack of mesitylene at the methene carbon concerted with the departure of the triazole anion (Scheme 1).

To explain the formation of (7) as the main product requires a competing process, since triazole could not be formed in a concerted process. One possibilty is that (2) undergoes N-N bond homolysis giving the triazolyl free radical (see later), which abstracts a benzylic hydrogen to give the observed (7), and a benzyl-type radical which will dimerize. Two facts argue against this proposal: there seems to be no precedent of such a homolysis taking place with the pyridiniums,1 and secondly, no products resulting from benzylic radical coupling were observed in this case. Had free (1c) been formed by N-N heterolysis, one would have expected at least some electrophilic attack on mesitylene to give a 1-triazolyl substitution product (as was observed in the diazonium ion decompositions). Since no such product was formed in the thermolyses, we suggest that the triazolyl cation forms a tight pair with collidine, which then undergoes a SET, leading to the collidine radical cation (25) and the triazolyl radical (26) (again within the tight pair). Abstraction of a hydrogen atom by triazolyl radical (26) from the 4-methyl group of (25) could lead to (7) and the 4-picolyl cation (27). Such benzyl-type cations are known to give diarylmethanes,12a,20 while the corresponding benzyl free radicals dimerize to give 1,2-diarylethanes.20 Thus, a Friedel-Crafts alkylation of mesitylene by (27) might account for the formation of (6). Hydride ion abstraction from (2) leading to (24), and formation of (6) from (24) will both lead to (7), as could other hydrogen-abstraction processes. That (4) and (5) (both 4-triazolyl compounds) but no 1-triazolyl compounds are formed in the thermolysis of the pyridiniums argues again in favor of a concerted mechanism leading to these products. On the other hand, (6) and (7) could well result from the heterolytic mechanism.
Photolyses.
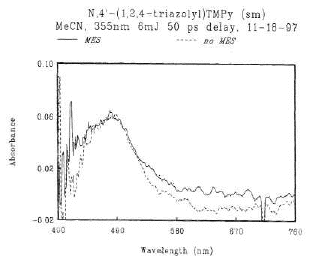
In contrast, the results from the photochemical reactions indicate that the products are probably formed by intermolecular stepwise processes. Some nitrenium ions have recently been detected using laser flash photolysis.13a,21 We have carried out very preliminary experiments on the laser flash photolysis (355nm, 6mJ) (30 ps pulse) of (2) and the corresponding 2,4,6-triphenylpyridinium salt. Both salts visibly fluoresce and show transients upon direct excitation. The absorbance maximum of the transient from (2) is at 485 nm (Fig. 4), and those from the triphenyl compound are at 450 and 630 nm (Fig. 5).
|
|
Fig. 4. Transient absorptions of (2) in the presence and absence of mesitylene. |
Fig. 5. Decay of transient absorptions of 1-(1,2,4-triazol-4-yl)-2,4,6-triphenylpyridinium tetrafluoroborate in mesitylene/acetonitrile (100 to 1000 ps). |
The transient from (2) decays on the timescale of a few nanoseconds, which decay is somewhat accelerated by the addition of mesitylene. The position and shape of the spectral band, shown in Fig. 4, is not altered by the presence of mesitylene. The spectrum arising from the triphenyl compound changes between 50 and 500 ps following the laser pulse, with the 450 nm peak decaying and producing a new absorption between 490 and 600 nm.
Owing to the observed lifetimes, the visible fluorescence, and the fact that that the two salts give different transients, we suggest that the bands at 485 and 450 nm could arise from the first excited singlet states of each of the pyridinium salts, rather than from an intermediate triazolyl cation. Indeed, triazole itself does not absorb to any appreciable extent above 200 nm. Falvey and Cramer13a, subjected N-(diphenylamino)-2,4,6-trimethylpyridinium tetrafluoroborate to LFP (308 nm) and observed two transients at 425 and 660 nm which they attributed to the singlet state of Ph2N+. This decayed to give bands at 325 and 680 nm, attributed to Ph2N+. .21 The latter species was generated by 266 nm LFP of N-nitrosodiphenylamine in the presence of malonic acid in acetonitrile solution and monitored at 690 nm -- the aminyl radical was formed first22 which then underwent protonation. The fact that only one major transient was observed for the trimethylpyridinium and two for the triphenylpyridinium could be interpreted speculatively as follows: the transients in the 450 - 485 nm range are owing to the singlet excitation; the one at 630 nm, observed only with the triphenylpyridinium, could be owing to the formation of an intermediate in the intramolecular attack of the 2-phenyl group by the triazolyl ring to give a diradical cation
s-complex (28) as sketched in Scheme 2.
Some credibility is lent to this hypothesis by the finding by Katritzky and coworkers that 1,2-diphenylpyridinium perchlorates undergo intramolecular ring-closure on irradiation with 300 nm light,23 and 2-phenyl-1-(1,2,3-triazol-4-yl)pyridinium salts undergo similar intramolecular cyclizations.23 The above results, along with the reported photochemical properties of pyridinium compounds in the presence of electron donors,24 led us to examine the possibility of a photoinduced electron transfer (PET) from mesitylene to salt (2). The intensity of fluorescence of (2) as a function of mesitylene concentration was measured and no change was observed in the shape, but a change was observed in the intensity of the fluorescence leading to the Stern-Volmer relationship24 displayed in Figs. 6a,b. The quenching rate constant, kq (= Kqt-1), in acetonitrile, obtained from the slope (Kq) of the Stern-Volmer plot and the fluorescence lifetime t (ca. 2 ns, from the LFP studies), is 1.4 x 1010 M-1s-1. A similar value (1.3 x 1010 M-1s-1) has been reported for electron transfer from mesitylene to 10-methylacridinium ion.25 Such an electron transfer would produce the mesitylene radical cation (29) and the pyridinyl radical (30), which could fragment to the triazolyl radical (26) and then lead to (10) (Scheme 3), as suggested for the formation of (4) in Scheme 1 above.
|
|
Fig. 6a. Fluorescence intensity of (2) as a function of the mesitylene concentration. |
Fig. 6b. Stern-Volmer plot of fluorescence of 2 in mesitylene/acetonitrile. |
Pyridinyl radicals, which are accessible by electrochemical reduction, photolysis,26a or by chemical means,26b are isolable in some cases,26c but usually dimerize or fragment. The latter is the predominant reaction observed in cases in which a heteroatom is bound to the pyridinyl nitrogen,26c such as is (30). To test if (30) could be a possible intermediate in our reactions, (2) was reduced with Na/Hg under nitrogen in acetonitrile solution.27 2,4,6-Collidine and triazole (7) were the major products, together with lesser amounts of collidine dimers. No detectable amounts of the dimer of radical (30) were detectable (NMR, GC/MS). This fragmentation of (30), together with the LFP spectroscopic data, strongly suggest that (10) is not formed via the intermediacy of (1).

On the other hand, the formation of (1c) as a tight pair with either 2,4,6-collidine or 2,4,6-triphenylpyridine may be required to explain to account for the formation of (13) (with traces of (4)) from (2) in the absence of mesitylene (Scheme 4). In the presence of added 2,6-dimethylpyridine this tight pair may be solvent-separated leading to the formation of (18) and (7) ((13) could still arise from the tight pair). A similar separation of the pair from the triphenylpyridinium in the presence of external collidine would explain the products similarly (here, (13) would necessarily arise from the solvent separated pair).

That no product resulting from hydrogen-abstraction from the pyridine side-chains was observed may be due to the fact that, if this abstraction did occur, it would produce a picolyl cation in which the positive charge would be delocalized over the nitrogen atom (i.e. a nitrenium ion) -- see argument above in the discussion of the thermal formation of (6).
The thermolysis of the diazonium salts from (3) and from (11) produces 77% of the parent triazole (7), a result consistent with the formation of the diradical cation (1c) (capable of hydrogen abstractions to form triazolium tetrafluoroborate, which provides (7) upon isolation). Also formed was N2O, which is consistent with either the formation of a hydroxyazo intermediate28 or a labile N-nitrosoamine,29 and the mesitylene dimer (12). The triazolomesitylenes (10) and (8) are formed from both (3) or (11) (in very low yield) in about the same ratio. This suggests the formation of a common intermediate. If mesitylene were reacting directly with the two diazonium salts (3) and (11) via an SN2' process, different ratios of (10) to (8) would be expected to be formed from the two aminotriazoles, so this can be excluded. That the reactive intermediate from the thermolysis is not a free radical30 was established by the photolysis of (21) (which gives rise to the radical) in the presence of mesitylene, but no (10) was formed in this way. The 1- and 4-triazolyl cations could produce (10) via electrophilic attack of mesitylene or via an SET process similar to that suggested for the thermolysis of (2). The hydrogen-abstraction product (7) must then arise from the triplet state (1c) of the diradical cation, which is predicted to be the ground state of (1).
Scheme 5
Unlike other nitrenium ions (singlets) generated photolytically from the corresponding pyridiniums,2e,14a the triplet diradical cation (1c) does not react in the same way with electron rich donors, but gives mainly the hydrogen abstraction product from suitable donors, as is typical of triplet nitrenium ions. Note in Table 1 the increase in yield of triazole (7) from 6% to 42% as the methanol-d4: acetonitrile ratio increases. In conclusion, therefore, it appears that the thermolysis of the pyridinium salt (2) does not generate free (1) and the products (with the possible exception of triazole itself) result mostly from concerted processes. Thermolysis of the N-diazonium salts produces (1), while photolysis of the pyridiniums in the presence of mesitylene gives products whose formation is best accounted by single electron transfer from the substrate to the pyridinum, whereas photolysis in methanol and acetonitrile is best explained by involving the intermediacy of (1). Finally, the triazolium's behavior (when it is formed) is well-accounted for by its predicted diradical cation structure (1c).
Acknowledgments.
This research was supported in part by the Spanish DGICYT (Grant PB98-1240-C02-01) and the Catalonian CIRIT (Grant 1999SGR-00043). HHG is grateful for the support from Austin
College and The Robert A. Welch Foundation. A grant from the National Science Foundation (CHE-9012972) to RAA is also gratefully acknowledged. We thank Reilly Industries for the gift of 4-amino-1,2,4-triazole. Dr. Mike Bockman carried out the laser flash photolysis experiments at the University of Houston under the direction of Professor Jay Kochi. Prof. Sun YaPing (Clemson) carried out the fluorescence studies. Their assistance is gratefully acknowledged.References and Notes
(1) Abramovitch, R. A.; Davis, B. A. Chem. Rev. 1964, 64, 149. Gassman, P. G. Acc. Chem. Res. 1970, 3, 26. Abramovitch, R. A. �Nitrenes� in �Organic Reactive Intermediates,� (McManus, Ed.), p. 127, Academic Press, 1973. Abramovitch, R. A.; Jeyaraman, R. �Nitrenium Ions,� in �Azides and Nitrenes,� (Scriven, E. F. V., Ed.), p. 297, Academic Press, 1984. Scriven, E. F. V.; Turnbull, K. Chem. Rev. 1988,88, 297. Simonova, T. P.; Nefedov, V. D.; Toropova, M. A.;Kirillow, N. F. Russian Chem. Rev. 1992, 61, 584, among others.(2) (a) Abramovitch, R. A.; Cooper, M.; Iyer, R.; Jeyaraman, R. Rodrigues, J. A. R. J. Org. Chem. 1982, 47, 4819. (b) Abramovitch, R. A.; Chinnasamy, P.; Evertz, K. Huttner, G. J. Chem. Soc., Chem. Commun. 1989,3. (c) deSousa, J. D. F.; Rodrigues, J. A. R.; Abramovitch, R. A. J. Am. Chem. Soc. 1994, 116, 9745. (d) Abramovitch, R. A.; Ye, X-C.; Pennington, W. T.; Schimek, G.; Bogdal, D. J. Org. Chem. 2000, 65, 343. (e) Takeuchi, H. J. Chem, Soc., Chem. Commun. 1987, 961.
(3) Novak, M.; Xu, L.L.; Wolf, R. A. J. Am. Chem. Soc. 1998, 120, 1643. Novak, M.; Kennedy, S. S. 1995, 117, 574. Humphreys, W. G.; Kadlubar, F. F.; Guengerich, F. P. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 8278. Colvin, M. E.; Nilsen, I. B. M.; LeBui,L.; Hatch, F. T. Chem. Biol. Interact. 1992, 81, 19, and references cited therein.
(4) (a) Abramovitch, R. A.; Evertz, G.; Gibson, H. H., Jr.; Weems, H. G., Jr. J.Chem. Soc., Chem. Commun 1988, 325. (b) Abramovitch, R. A.; Beckert, J. M.; Chinnasamy, P.; He, X-H.; Pennington, W. T.; Sanjivamurthy, A. R. V. Heterocycles 1989, 28, 623
(5) Rutchenko, V.; Ignatov, S.; Kostyanovsky, R. J. Chem. Soc., Chem. Commun. 1990, 261.
(6) (a) Abramovitch, R. A.; Beckert, J. M.; Pennington, W. T. J. Chem. Soc., Perkin Trans. 1 1991, 761. (b) Gilchrist, T. L.; Rees, C. W.; Thomas, C. ibid. 1975, 12.
(7) Fabian, J.; Melhon, A.; Tynkylkov, N. Theochem. 1987, 151, 355.
(8) Gompper, R.; Junius, M. Terahedron Lett. 1980, 21, 2883. Gompper, R.; Guggenberger, R.; Zantgraf, R. Angew. Chem. Int. Ed. Engl. 1985, 24, 984.
(9) Abramovitch, R. A.; Gibson, H. H.; Nguyen, T.; Olivella, S.: Sole, A. Tetrahedron Lett. 1994, 35, 2321.
(10) Li, Y.; Abramovitch, R. A.; Houk, K. J. Org. Chem. 1989, 54, 2911. Ford, G. P.; Herman, P. S. J. Chem. Soc., Perkin. Trans. 2 1991, 616. Smith, D. A.; Bitar, J. J. Org. Chem. 1993, 58, 6.
(11) Grundmann, C.; Ratz, R. J. Org. Chem. 1956, 21, 1037.
(12) (a) Wen, L.-S.; Kovacic, P. Tetrahedron 1978, 34, 2723. Kovacic, P.; Wu, C. J. Org. Chem. 1961, 26, 749. (b) See, however, Takeuchi, H. J. Chem. Soc., Chem. Commun. 1987, 961, and discussion in footnote to ref. 4a above.
(13) (a) Moran, R. J.; Cramer, C;, Falvey, D. E. J. Org. Chem. 1997, 62, 2742. (b) The known reactivity of acetonitrile towards cations, radicals and radical cations may explain, in part, the low material balance in some of our reactions, although we did not detect any Ritter reaction products or stable solvent-derived products. See Lisjer, H. J. P.; Arnold, D. R. J. Phys. Chem. A 1998, 102, 5592.
(14) Fleming, I.; Dunogues, J.; Smithers, Organic Reactions 1989, 37?57.
(15) Ohga, K.; Yoon, U. C.; Mariano, P. S. J. Org. Chem. 1984, 49, 213.
(16) The low yield of 19 may also be owing to the reported desilylation of the allylsilane-derived radical cation by the weakly nucleophilic acetonitrile. Ohga, K.; Mariano, P. S. J. Amer. Chem. Soc. 1982, 104, 617.
(17) See the similar generation of the 1,2,3-triazolyl radical. Kalambokis, E. A.; Maroulis, A. J.; Alexandrou, N. E. J. Heterocycl. Chem. 1993, 30, 1301.
(18) t-Butyl 1-(1,2,4-triazolepercarboxylate), synthesized from t-butylhydroperoxide and 1,1'-carbonylbis-(1,2,4-triazole), underwent nucleophilic attack at the carbonyl group, as does t-butyl 1-imidazolepercarboxylate. Bourgeois, M. J.; Filliatre, C.; Lalandem R.; Maillard, B.; Villenave, J. J. Tetrahedron Lett. 1978, 36, 3355.
(19) In a previous study4 of the low-lying electronic states of 1, it was found that the optimum C2
n structure calculated at the CASSCF(10,8)/6-31G(d) level for the six p electron singlet state 1A1 showed one negative vibrational frequency of b2 symmetry. This spurious result was a consequence of the fact that the force constant matrix was calculated numerically by finite differences of analytical gradients rather than from the analytical second derivatives of the energy.(20) Kozhevnikov, I. V.; Kim, V. I.; Talzi, E. P.; Sidelnikov, V. N. J. Chem. Soc., Chem. Commun. 1985, 1392. Eberson, L.; Nyberg, K. Accts. Chem. Res. 1973, 6, 106. Abramovitch, R. A.; Roy, J.; Uma, V. Can. J. Chem. 1965, 43, 3407.
(21) (a) Moran, R. J.; Falvey, D. E. J. Am. Chem. Soc. 1996, 118, 8965. (b) Chiapprino, D.; Falvey, D. E. J. Phys. Org. Chem. 1997, 10, 917. (c) Srivastava, S.; Kercher, M.; Falvey, D. E. J. Org. Chem. 1999, 64, 5833.
(22) Wagner, B. D.; Ruel, G.; Lusztyk, J. J. Amer. Chem. Soc. 1996, 118, 13.
(23) (a) Katritzky, A. R.; Zakaria, Z.; Lunt, E. J. Chem. Soc., Perkin Trans.1 1980, 1879. (b) Katritzky, A. R.; Agha, B.; deVille, G. Z. Org. Mag. Resonance 1983, 21, 649.
(24) (a) Mariano, P. Accts. Chem. Res. 1983, 16, 130. (b) See also: Bockman, T. M.; Lee, K.H.; Kochi, J.; J. Chem. Soc., Perkin Trans. 2 1992, 1581.
(25) Fujita, M.; Ishida, A.; Takamuku, S.; Fukuzumi, S. J. Amer. Chem. Soc. 1996, 118, 8566.
(26) (a) Kosower, E.M.; Schwinger, I. J. Amer. Chem. Soc. 1964, 86, 4493. (b) Kosower, E. M. �Molecular Biochemistry,� McGraw Hill, N.Y. 1962. (c) Kosower, E. M.. "Free Radicals in Biology," (Pryor, W. A., Ed.), Vol. II, Ch. 1, Academic Press, New York, 1976.
(27) Kosower, E. M.; Ikegami, Y. J. Amer. Chem. Soc. 1967, 89, 461.
(28) (a) Kreher, R.; Bergmann, V. Z. Naturforsch., Anorg. Chem., Org. Chem. 1992, 37b, 881; (b) DeRosa, M.; Haberfield, P. J. Org. Chem. 1981, 46, 2639, and references cited therein.
(29) Kuzmenko, V. V.; Pozharskii, A. F. Adv. Heterocycl. Chem. 1992, 53, 154.
(30) March, J. �Advanced Organic Chemistry, Reactions, Mechanisms, and Structure, 4th Ed.,� Ch. 14, Sec. 4-18, John Wiley & Sons, New York, 1992.
All comments on this poster should be sent by e-mail to [email protected] with A0019 as the message subject of your e-mail.