There are few general methods to reduce the nitrogen-nitrogen bond in hydrazines, and the conditions required to cleave this bond depend markedly on the substituents attached to it [6]. However, substituted hydrazines can be reduced to the corresponding amines by catalytic hydrogenation [7] or using metals in a protic solvent [8, 9]. In the first case, usually it is necessary to work under pressure and acidic medium, and in the second case, liquid ammonia is the most common solvent.
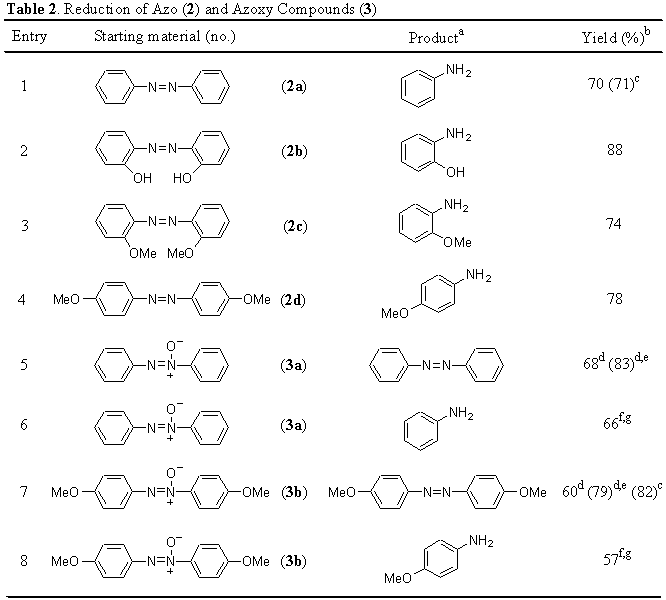
The reaction of monosubstituted, N,N- or N,N�-disubstituted hydrazines 1 with an equimolar amount of dihydrated nickel chloride, an excess of lithium powder [8:1 molar ratio, referred to the nickel(II) salt] and a catalytic amount of 4,4�-di-tert-butylbiphenyl (DTBB) [0.1:1 molar ratio, referred to the nickel(II) salt] in THF at room temperature overnight, led to the formation of the corresponding primary or secondary amines (Table 1).
a
All products were >95% pure (GLC). b Isolated yield after acid-base extraction. c Reaction performed in the presence of a vinylbiphenyl-divinylbenzene copolymer as electron carrier. d Yield corresponding to the reaction: 1f � 2PhNH2.
As an alternative for the activation of nickel in the reduction of hydrazines 1, it is also possible to use a polymer supported arene as the catalytic electron transfer agent. The preparation of a polymer containing biphenyl was carried out following Itsuno�s methodology [10] to give cross-coupling polymers. Thus, by refluxing a solution of 4-vinylbiphenyl and divinylbenzene in tetrahydrofuran, containing a catalytic amount of azobis(isobutyronitrile), the corresponding polymeric catalyst was obtained (Scheme 1). Using 4-vinylbiphenyl-divinylbenzene copolymer [4], a slightly better yield was obtained for the reduction of N-methyl-N-phenylhydrazine (1d) (Table 1, entry 4 and footnote c).
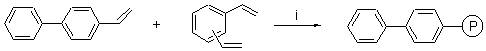
Scheme 1. Reagents and conditions: i, AIBN, THF, reflux
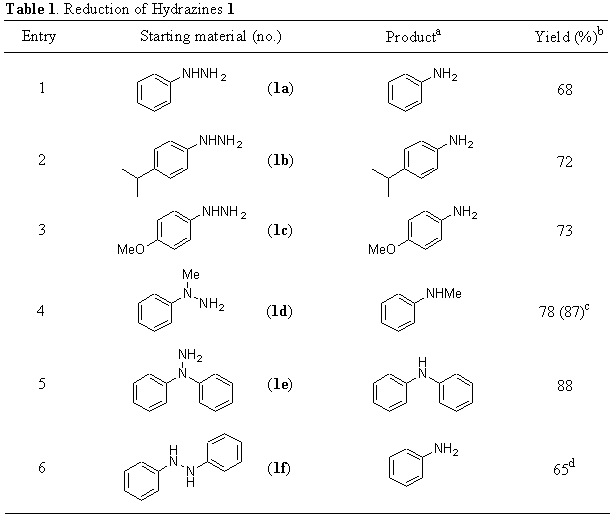
The most important methods to reduce azo compounds lead to the corresponding hydrazo derivatives [11]. They include diimide, complex metal hydrides or cobalt boride in the presence of hydrazine [12], as well as metal mediated procedures [13, 14]. Concerning azoxy compounds, the corresponding deoxygenation is the most studied reaction [15]: well-established methods include catalytic hydrogenation [16] and metal-mediated procedures [17]. In addition, azoxy compounds are cleaved to amines by potassium borohydride an copper(I) chloride [18].
The application of the procedure described above for hydrazines 1 was also applied to azo compounds 2, leading to the corresponding primary aromatic amines (Table 2, entries 1-4). In the case of azoxy derivatives 3, the final products depend on the reaction conditions used: for short reactions times (ca. 1h), the corresponding azo compounds were isolated (Table 2, entries 5 and 7, and footnote d), whereas either longer reaction time (10 h) or 2.5 eq. of dihydrated nickel(II) chloride led to full reduction to the corresponding primary amines (Table 2, entries 6 and 8, and footnotes f and g). Also here, the use of an arene-supported polymer as electron carrier catalyst [4] in the reaction showed to be as effective as the use of DTBB (Table 2, entries 1 and 7, and footnote c).