Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0021]
Arene-Catalysed Reductive Cleavage of the Benzylic Carbon-Sulfur Bond: Generation of Benzylic Lithium Reagents.
E. Alonso, J. V. Ferrandez, F. Foubelo, I. Gomez, D. Guijarro, A. Gutierrez, P. J. Martinez, O. Prieto, D. J. Ramon, T. Soler and M. Yus*
Departamento de Quimica Organica, Universidad de Alicante, E-03080 Alicante, Spain.
Tel. +34-965903548, Fax +34-965903549, E-mail: [email protected], http://www.ua.es/dept.quimorg/
With one biographical summary
Received: 18 July 2000 / Uploaded: 29 July 2000
Abstract: The reductive cleavage of benzylic carbon-sulfur bonds via an arene-catalysed lithiation process is described. The benzyllithium reagents generated were reacted with several electrophiles, mainly carbonyl compounds, affording the expected products in moderate to good yields.
Keywords: arene-catalysed lithiation, benzyllithium, carbon-sulfur bond cleavage.
Introduction
Typical methods for the preparation of organolithium reagents involve halogenated derivatives as starting materials. However, this general procedure is not useful for the generation of allylic or benzylic lithium intermediates, due to the almost exclusive formation of Wurtz-type products [1-3]. Other alternative methodologies have been reported, such as: (a) the direct deprotonation of the corresponding allylic or benzylic hydrocarbons with an alkyllithium, which needs the use of a co-reactant such as an alkoxide [4,5] or an amine [6]; (b) a mercury-lithium or tin-lithium transmetallation [7]; (c) the reductive cleavage of allyl phenyl ether with lithium, which has been successfully used in the case of allyllithium [8,9].
On the other hand, in recent years we have used an arene-catalysed lithiation [10,11] of different substrates [12-14] in order to prepare organolithium reagents under very mild reaction conditions. In this communication we describe the application of the arene-catalysed lithiation to the generation of benzylic organolithium compounds starting from 1,7-dihydrodibenzothiepin and benzyl mercaptan, via a carbon-sulfur reductive cleavage.
Results and Discussion
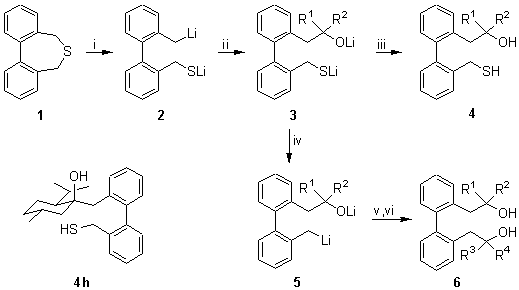
The reaction of 1,7-dihydrodibenzothiepin (1) with an excess of lithium powder and a catalytic amount of 4,4�-di-tert-butylbiphenyl (DTBB, 5 mol%) in THF at �78deg.C for 1 h, followed by treatment with a carbonyl compound at the same temperature and final hydrolysis with water, led to the formation of the corresponding sulfanyl alcohols 4 in good yields (Scheme 1 and Table 1, entries 1-8), dianionic intermediates 2 and 3 being involved in this process. When, after addition of the carbonyl compound, the system was allowed to reach room temperature, carbon-sulfur reductive cleavage took place to give oxygen-functionalised benzylic organolithium compounds 5, which, by reaction with a second carbonyl compound at �78deg.C and final hydrolysis with water at the same temperature, gave the corresponding diols 6 in moderate yields (Scheme 1 and Table 1, entries 9-10).
Scheme 1. Reagents and conditions: i, Li, DTBB (5 mol %), THF, -78deg.C, 1 h; ii, E1+= R1R2CO, -78deg.C, 5 min; iii, H2O-HCl, -78 to 20deg.C, 3 h; iv, -78 to 20deg.C, 1 h; v, E2+= R3R4CO, -78deg.C, 5 min; vi, H2O, -78 to 20deg.C, 3 h.
Table 1. Preparation of Compounds 4 and 6 from 1,7-Dihydrodibenzothiepin
Producta |
||||||
Entry |
E1+ |
E2+ |
No. |
R1R2COH |
R3R4COH |
Yield (%)b |
1 |
ButCHO |
--- |
4a |
ButCHOH |
--- |
71 |
2 |
Ph(CH2)2CHO |
--- |
4b |
Ph(CH2)2CHOH |
--- |
75 |
3 |
PhCHO |
--- |
4c |
PhCHOH |
--- |
82 |
4 |
Me2CO |
--- |
4d |
Me2COH |
--- |
50 |
5 |
[CH3(CH2)4]2CO |
--- |
4e |
[CH3(CH2)4]2COH |
--- |
76 |
6 |
(CH2)5CO |
--- |
4f |
(CH2)5COH |
--- |
72 |
7 |
(CH2)7CO |
--- |
4g |
(CH2)7COH |
--- |
47 |
8 |
(-)-menthone |
--- |
4hc |
--- |
--- |
55 |
9 |
(CH2)5CO |
(CH2)5CO |
6a |
(CH2)5COH |
(CH2)5COH |
30 |
10 |
(CH2)5CO |
Me2CO |
6b |
(CH2)5COH |
Me2COH |
45 |
a
All products were >95% pure (GLC and 300 MHz 1H NMR). b Isolated yield after column chromatography (silica gel, hexane/ethyl acetate). c See Scheme 1The naphthalene catalysed lithiation process was also applied to the conversion of benzyl mercaptan 7 into benzyllithium. Compound 7 was first reacted with n-butyllithium in order to remove the acidic proton on sulfur. The resulting thiolate was then treated with lithium powder (1:10 molar ratio) and a catalytic amount of naphthalene (1:0.20 molar ratio, 10 mol %) in THF at 0deg.C, leading to a solution of benzyllithium, which reacted with several electrophiles (1:1.2 molar ratio) at temperatures ranging between -30 and 0deg.C, affording the expected products 8 after hydrolysis with water (Scheme 2 and Table 2).
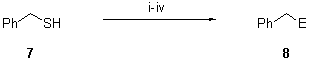
Scheme 2. Reagents and conditions: i, n-BuLi, THF, 20deg.C, 5 min; ii, Li, C10H8 (10 mol %), THF, 0deg.C, 30 min; iii, E+ = t-BuCHO, PhCHO, Et2CO, (CH2)5CO, Me3SiCl, -30 to 0deg.C, 30 min; iv, H2O.
Table 2. Preparation of Compounds 8 from Benzyl Mercaptan.
Producta |
||||
Entry |
E+ |
No. |
E |
Yield (%)b |
1 |
ButCHO |
8a |
ButCHOH |
48 |
2 |
PhCHO |
8b |
PhCHOH |
52 |
3 |
Et2CO |
8c |
Et2COH |
43 |
4 |
(CH2)5CO |
8d |
(CH2)5COH |
45 |
5 |
Me3SiCl |
8e |
Me3Si |
46 |
a
All products were >95% pure (GLC and 300 MHz 1H NMR). b Isolated yield after column chromatography (silica gel, hexane/ethyl acetate).
Removal of the acidic proton on sulfur was necessary in order to get good yields. Almost no product was formed when the reaction was carried out without previous deprotonation. On the other hand, yields were better when the electrophile was added at -30deg.C and the reaction was stirred allowing the temperature to rise to 0deg.C in ca. 30 min. When the electrophile was added at the same temperature of the lithiation process (0deg.C), yields of the expected products varied from 25 to 30%, mainly due to the formation of the corresponding pinacol-type products as a side reaction, in the case of using a carbonylic compound as electrophilic reagent. Cooling the reaction mixture to -30deg.C before the addition of the electrophile, the formation of those by-products was diminished.
Conclusion
Reductive cleavage of benzylic carbon-sulfur bond in 1,7-dihydrodibenzothiepin led, after reaction with carbonyl compounds, to sulfanyl alcohols or, depending on the reaction conditions, to diols, acting in this case the thiepin derivative as precursor of a synthetic equivalent of 2,2�-bis(lithiomethyl)biphenyl. On the other hand, benzyl mercaptan could be used as a source of benzyllithium through a naphthalene catalysed process, via the same kind of reductive cleavage.
Experimental Part
For general information, see reference [15].
Preparation of 2-(2-Hydroxy-2-methylpropyl)-2�-(sulfanylmethyl)biphenyl (4d). Typical Procedure.
To a cooled (-78deg.C) blue suspension of lithium powder (100 mg, 14.0 mmol) and a catalytic amount of 4,4�-di-tert-butylbiphenyl (40 mg, 0.15 mmol) in THF (5 ml) was added dropwise a THF solution (1 ml) of 1,7-dihydrodibenzothiepin (212 mg, 1.0 mmol) under nitrogen and the mixture was stirred at the same temperature for 1 h. Then, acetone (64 mg, 0.08 ml, 1.1 mmol) was added at �78deg.C and after 5 min the reaction mixture was hydrolysed with 1M hydrochloric acid (20 ml) and extracted with ethyl acetate (3x20 ml). The organic layer was dried over anhydrous sodium sulfate and evaporated (15 Torr). The resulting residue was purified by column chromatography (silica gel, hexane/ethyl acetate) to yield pure compound 4d (136 mg, 50%).
IR (film): 3420-3230 (OH), 3090 (Ar-H).
1
H-NMR (CDCl3): 0.99 (s, 3H), 1.06 (s, 3H), 1.59 (t, 1H), 2.55 (d, 1H), 2.76 (d, 1H), 3.12-3.21 (br s, 1H), 3.47 (dd, 2H), 7.14-7.47 (m, 8H).13
C-NMR (CDCl3): 26.4, 29.3, 29.8, 45.5, 71.5, 126.3, 126.6, 127.3, 127.85, 128.9, 130.4, 130.75, 131.0, 135.8, 138.9, 140.4, 140.8.MS (EI): 254 (M+-H2O, 1%), 215 (11), 214 (64), 205 (12), 166 (28), 165 (65), 152 (12), 59 (100), 43 (28).
Preparation of 2,2�-Bis(1-hydroxycyclohexylmethyl)biphenyl (6b). Typical Procedure.
To a cooled (-78deg.C) blue suspension of lithium powder (100 mg, 14.0 mmol) and a catalytic amount of 4,4�-di-tert-butylbiphenyl (40 mg, 0.15 mmol) in THF (5 ml) was added dropwise a THF solution (1 ml) of 1,7-dihydrodibenzothiepin (212 mg, 1.0 mmol) under nitrogen and the mixture was stirred at the same temperature for 1 h. Then cyclohexanone (109 mg, 0.120 ml, 1.1 mmol) was added at �78deg.C and after 5 min the cold bath was removed and stirring was continued at 20deg.C for 1.5 h. After that, the reaction mixture was cooled down at �78deg.C and cyclohexanone (109 mg, 0.120 ml, 1.1 mmol) was added dropwise and the temperature was allowed to rise to 20deg.C overnight. The resulting mixture was hydrolysed with water (20 ml) and extracted with ethyl acetate (3x20 ml). The organic layer was dried over anhydrous sodium sulfate and evaporated (15 Torr). The resulting residue was purified by column chromatography (silica gel, hexane/ethyl acetate) to yield pure compound 6b (170 mg, 45%).
IR (film): 3500-3120 (OH), 3060 (Ar-H).
1
H-NMR (CDCl3): 0.92-1.47 (m, 22H), 2.57 (d, 2H), 2.63 (d, 2H), 7.15-7.41 (m, 8H).13
C-NMR (CDCl3): 21.7, 21.9, 25.5, 27.9, 72.0, 126.3, 126.85, 131.0, 131.2, 135.1, 142.5.MS (EI): 360 (M+-H2O, 2%), 265 (13), 263 (62), 254 (21), 246 (38), 215 (25), 184 (12), 59 (100), 43 (18).
Preparation of 3,3-Dimethyl-1-phenyl-2-butanol (8a). Typical Procedure.
To a solution of benzyl mercaptan (0.12 ml, 1.0 mmol) in THF (2 ml), n-butyllithium (0.63 ml 1.6M solution in hexane, 1.0 mmol) was added at 20deg.C. After 5 min, the resulting solution was slowly added (ca. 30 min) via syringe to a green suspension of lithium powder (70 mg, 10.0 mmol) and naphthalene (26 mg, 0.2 mmol) in THF (5 ml), under nitrogen, at 0deg.C. After 30 min stirring at the same temperature, the reaction mixture was cooled to -30deg.C and pivalaldehyde (0.13 ml, 1.2 mmol) was added and the reaction was stirred allowing the temperature to rise to 0deg.C (ca. 30 min). The reaction was then hydrolysed with water (10 ml), acidified with 2M hydrochloric acid (6 ml) and extracted with ethyl acetate (3x20ml). The combined organic layers were washed with saturated sodium bicarbonate (5 ml), water (5 ml) and brine (5 ml) and then dried over anhydrous sodium sulfate. Solvents were evaporated (15Torr) and the resulting residue was purified by column chromatography, affording pure compound 8a (85 mg, 48%).
IR (oil): 3472 (OH), 3084, 3062, 3027, 1604, 1494 (HC=C).
1
H-NMR (CDCl3): 0.99 (s, 9H), 2.46 (dd, 1H), 2.90 (dd, 1H), 3.42 (dd, 1H), 7.19-7.33 (m, 5H).13
C-NMR (CDCl3): 25.85, 38.35, 77.0, 80.55, 126.25, 128.55, 129.3, 139.9.MS (EI): 178 (M+, <1%), 121 (18), 103 (17), 92 (100), 87 (25), 69 (26), 65 (16), 57 (22), 45 (27), 43 (19), 41 (46).
Acknowledgements
The DGICYT of the Spanish Ministerio de Educacion y Cultura (no. PB97-0133) and the Generalitat Valenciana (no. GV-C-CN-09-066-96 and DGDOC99-02-4) generously supported the work presented here.
References and Notes
[1] Wakefield, B. In Comprehensive Organic Chemistry; Barton, D.; Ollis, V. D. Eds.; Pergamon Press: Oxford, 1979; vol.3, p. 414.
[2] Negishi, E.-I. In Organometallics in Organic Synthesis; J. Wiley & Sons: New York, 1980; p. 96.
[3] Wakefield, B. In Organolithium Methods; Academic Press: London, 1988; p. 38.
[4] Lochmann, L.; Pospisil, J.; Lim, D. Tetrahedron Lett. 1966, 257.
[5] Schlosser, M. J. Organomet. Chem. 1967, 8, 9.
[6] Akiyama, S.; Hooz, J. Tetrahedron Lett. 1973, 4115.
[7] Schlosser, M. In Organometallics in Organic Synthesis; J. Wiley & Sons: Chichester, 1944; p. 53.
[8] Eisch, J. J.; Jacobs, A. M. J. Org. Chem. 1963, 28, 2145.
[9] Eisch, J. J. Organomet. Synth. 1981, 2, 91.
[10] Yus, M. Chem. Soc. Rev. 1996, 155.
[11] Ramon, D. J.; Yus, M. Eur. J. Org. Chem. 2000, 225.
[12] Foubelo, F.; Yus, M. Rev. Heteroatom. Chem. 1997, 17, 73.
[13] Foubelo, F.; Yus, M. Trends Org. Chem. 1998, 7, 1.
[14] Guijarro, D.; Yus, M. Recent Res. Devel. Org. Chem. 1998, 2, 713.
[15] Ramon, D. J.; Yus, M. Tetrahedron: Asymmetry 1997, 8, 2479.
Miguel Yus was born in Zaragoza (Spain) in 1947, and received his BSc (1969), MSc (1971) and PhD (1973) degrees from the University of Zaragoza. After spending two years as a postdoctoral fellow at the Max Planck Institut fur Kohlenforschung in Mulheim a.d. Ruhr he returned to Spain to the University of Oviedo where he became assistant professor in 1977, being promoted to full professor in 1987 at the same university. In 1988 he moved to a chair in Organic Chemistry at the University of Alicante where he is currently the head of the Organic Chemistry Department. Professor Yus has been visiting professor at different institutions and universities such as ETH-Zentrum, Oxford, Harvard, Uppsala, Marseille and Tucson. He is a member or fellow of the chemical societies of Argentina, UK, Germany, Japan, Spain, Switzerland and USA. Among other awards, Professor Yus received in 1999 the "JSPS Prize" and the "Prix Franco-Espagnol" from the Japanese and French Chemical Societies, respectively. He is co-author of about 250 papers mainly in the field of development of new methodologies involving organometallic intermediates. His current research interest is focused on the preparation of very reactive functionalised organometallic compounds and their use in synthetic organic chemistry, arene-catalysed activation of different metals and preparation of new metal-based catalysts for homogeneous and hetereogeneous selective reactions.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0021 as the message subject of your e-mail.