
Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0029]
Synthesis And Electrophilic Substitution of Pyrido[2,3,4-kl]acridines
Avi Koller+, Amira Rudi+, Marta Garcia Gravalos‡ and Yoel Kashman+*
+ School of Chemistry, Tel Aviv University, Ramat Aviv 69978,
Israel
E-mail: [email protected]
‡
PharmaMar, Tres- Cantos, Madrid, SpainReceived: 22 July 2000 / Uploaded: 29 July 2000
Keywords: pyrido[2,3,4-kl]acridines / Biomimetic synthesis / NMR / Electrophilic nitration / Cytotoxicity
Several new pyrido[2,3,4-kl]acridines were synthesized by reacting naphthoquinone, juglone and cyclohexan-1,3-dione with b,b’-diaminoketones in a biomimetic reaction. The structure of all new compounds (6a, b, 8, 10, 11, 12, 14, 15, 16, 17a, b, 20, 21) was elucidated by NMR and MS spectroscopy. Elecrophilic substitution, mainly nitration, of the various compounds was undertaken and the substitution positions determined. A series of derivatives were prepared and their cytotoxicity towards P-388 mouse lymphoma cells analysed. The most cytotoxic derivatives were found to have IC50’s of 0.1 ug/ml. |
Introduction
Over the last 15 years more than 50 pyridoacridine alkaloids, based on the 4H-pyrido[2,3,4-kl]acridone (1) skeleton (Figure 1), have been isolated from marine organisms.[1]
Almost all natural pyridoacridines have been reported to possess significant cytotoxicity against cultured tumor cells[1]. This motivated us to synthesize and study some of the compounds of this group.
In 1993 we reported a biomimetic synthesis of the pyrido[2,3,4-kl]acridine ring system by the reaction of b,b’-diaminoketones with a variety of quinones and diketones.[2][3] Using this method we synthesized the marine alkaloid ascididemin (2)[4] eilatin (3)[3] (Figure 1) and also new pyridoacridine skeletons such as benzoascididemin and isoeilatin.[5]
Here we report a biomimetic synthesis of additional new pyridoacridines and a study of their reactions with electrophiles or amines (in the case of the quinoneimines). Most of the new pyridoacridines were tested for in-vitro activity against tumor cells and some of them were found to be highly cytotoxic.
Results and discussion
Several new pyridoacridines were synthesized in a two step reaction of b ,b ’-diaminoketones with quinones. Thus, addition of 2,2’-diaminobenzophenone 4a or 4b to 1,4-naphthoquinone afforded in the first step the arylaminonaphthoquinones 5a and 5b respectively, in approximately 50% yield (Scheme 1). The reaction took place in the presence of catalytic amounts of cerous chloride while air was bubbled through the solution to oxidize the intermediate hydroquinone.[6][7] In the second step, treatment of compounds 5a and 5b in methanol with NH4OH at room temperature for 7 days gave the appropriated compounds 6a and 6b in over 90% yield (Scheme 1).

Table 1. Long-range CH correlations observed in the HMBC experiments of the benzopyridoacridines
C# |
H# of correlated protons |
|||||||
6a |
6b |
10 |
12 |
13a |
14 |
17a |
17b |
|
1 |
3 |
3 |
3 |
3 |
3 |
|||
2 |
4 |
4, OMe |
4 |
4 |
4 |
4 |
4, OMe |
4, OMe |
3 |
1 |
1 |
1 |
1 |
1 |
1 |
||
4 |
2 |
2 |
2 |
2 |
2 |
|||
4a |
1 , 3 | 1 , 3 | 1 , 3 | 1 , 3 | 1 , 3 | 1 | ||
4b |
4 , 5 |
4 , 5 |
4 , 5 |
4 , 5 |
4 , 5 |
4 , 5 |
4 , 5 |
4 , 5 |
4c |
6 , 8 |
6 , 8 |
6 , 8 |
6 , 8 |
6 , 8 |
6 , 8 |
6 |
6 |
5 |
7 |
7 |
7 |
7 |
7 |
|||
6 |
8 |
8 |
8 |
8 |
8 |
8 |
||
7 |
5 |
5, OMe |
5 |
5 |
5 |
5 |
5, OMe |
5, OMe |
8 |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
8a |
5 , 7 |
5 |
5 , 7 |
5 , 7 |
5 , 7 |
5 , 7 |
5 |
5 |
9a |
||||||||
10 |
11 |
11 |
11 |
11 |
11 |
|||
10a |
12, 14 | 12, 14 | 12, 14 | 12, 14 | 12,14,OH | 12, 14 | 14 | 12 |
11 |
13 |
13 |
13 |
13 |
13 , OH |
13 |
13 |
13 |
12 |
14 |
14 |
14 |
14 |
14 , OH |
14 |
14 |
|
13 |
11 |
11 |
11 |
11 |
11 |
|||
14 |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
14a |
11, 13 |
11, 13 |
13 |
13 |
13 |
11, 13 |
11, 13 |
11, 13 |
14b |
14 |
14 |
14 |
14 |
14 |
14 |
14 |
|
14c |
||||||||
15a |
2 , 4 |
4 |
2 , 4 |
2 , 4 |
2 , 4 |
2 , 4 |
4 |
4 |
The structures of 6a and 6b, possessing the required molecular ions (m/z 332 and 392, respectively) were confirmed by 1D and mainly COSY, HMQC and HMBC 2D NMR spectra. (See Table 1 for the HMBC correlations). Characteristic were the resonances of C-10 and C-14b of the quinoneimine system (ring C) and the down-field proton resonances of the spatial close protons H-4 and H-5 (d H 9.09 and 9.18 ppm, respectively, for 6a and d H 8.90 and 8.98 ppm, respectively, for 6b).[5] Three four-spin systems were observed in the 1H-NMR spectrum of 6a belonging to rings A, E and F. Rings A and E, carrying the spatial close H-4 and H-5 protons, were distinguished from ring F by NOE measurements. The differentiation between rings A and E was achieved from an NOE between H-1 and H-14 (about 3.7 A apart). This NOE was also the key for determining the structure of the nitration products 21, 23a and 23b as described below.
A second experiment that was performed with naphthoquinone was its reaction with TFA-kynuramine (7)[4] (Scheme 1). This reaction afforded 9H-benzo[i]pyrido[2,3,4-kl]acridin-9-one (8), deazaascididemin, earlier synthesized by Zjawiony by a four step reaction.[8] The structure of compound 8 (m/z 282) was confirmed by its NMR data (Experimental) and comparison with the data in the literature.[8]
A second naphthoquinone tested was juglone. Reacting juglone (5-hydroxy-1,4-naphthoquinone) with diaminobenzophenone 4a afforded in a regioselective reaction a single addition product 9 in 80% yield (Scheme 2).
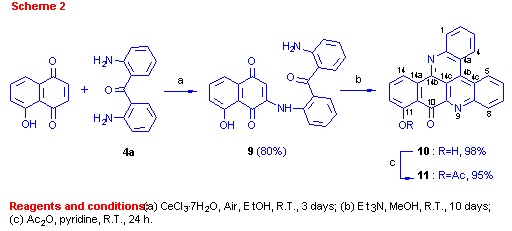
The orientation of this addition was defined by the structure determination of compound 10, obtained in the second step by stirring compound 9 in methanol with Et3N. A key HMBC correlation in the structure elucidation was the one between C-14b (d 147.5) and H-14 (d 8.73).
For other correlations that assisted with the structure determination see Table 1. The regioselectivity of nucleophilic additions of amines to juglone was observed previously by Thomson[9] in the reactions of aniline with the juglone derivatives 5-acetoxy or 5-methoxy-1,4-naphthoquinones.
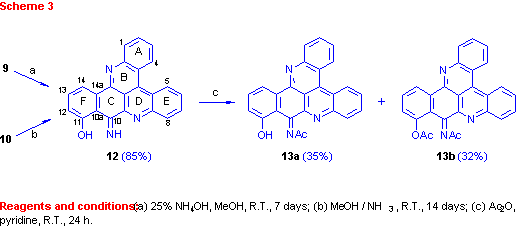
Performing the second step of the latter reaction with ammonia, rather than Et3N, as used for the preparation of compounds 6a and 6b, caused unexpectedly the disappearance of the C-10 carbonyl group. Moreover, acetylation of the obtained pyridoacridine (12) (Scheme 3) gave a mono- (13a) and a diacetate (13b). It is suggested that the carbonyl group of compound 10 is replaced in compound 12 by an imine and indeed, treatment of 10, obtained with the Et3N, with NH3 gave compound 12.
The position of the imine group was defined by a HMBC experiment of compound 13a namely from correlations between the 11-hydroxylic proton and carbons C-10a, C-11, and C-12 of ring F (see Table 1).
A major target in the present investigation was the study of the electrophilic substitution of pyridoacridines for the preparation of derivatives for structure activity relationship studies.
Investigating a variety of nitration conditions ( HNO3-TFA, HNO3-H2SO4 and NO2BF4 in CH3CN) brought to the best conditions, namely, the use of HNO3-H2SO4, 1:1 vide infra.
The nitration of compound 6a afforded a mono-nitro product 14 in 53% yield after 12 hours at room temperature. Because of the absence of long range CH-correlations in the NMR experiments between atoms of rings A or E and F to ring C it was difficult to determine whether the nitro group is attached to ring A or E. However, the nitration position, C-3 on ring A, could be determined from a NOE between H-1 and H-14 (2%), which are ca.3.7 A apart (see Scheme 4). It was found by 1D and 2D NMR experiments (for HMBC correlations see Table 1) that the nitration went to the para position of ring A.

Catalytic hydrogenation of compound 14 with 5% Pd-C in AcOH / TFA afforded the amino derivative 15.
Nitration of compound 6b, the electron richer 2,7-dimethoxy derivative of 6a, gave after 1 hour a dinitro derivative 16 and after 12 hours of reaction at room temperature two tetra nitro isomers 17a and 17b (Scheme 4). That the two nitro groups in 16 substituted C-1 and -8, ortho to the quinoline-nitrogen, was clear from the two AB- systems seen in the 1H-NMR spectrum along with the aromatic four- proton system.
The structures of 17a and 17b were also determined by 1D and 2D NMR experiments (for HMBC correlations see Table 1). In compounds 17a and 17b only one of rings A or E was attacked by the electrophile at the para position; their structures are tentatively suggested on the basis of the structure of compound 14 as depicted in Scheme 4. Because of the nitro groups at the ortho positions, it was impossible to prove by NOE that the substitution is at the para position of ring A (as in the case of compound 14). In addition to the nitration of rings A and E, ring F in 17a and 17b was also substituted due to long range activation by the methoxyl groups.
Oxidation of compounds 18 and 19 (obtained by condensation of compounds 4a and 4b with 1,3-cyclohexanedione) with cerium ammonium nitrate afforded benzopyridoacridones 20 and 21, respectively, in high yields (Scheme 5). Amination of the latter quinoacridones (20 and 21) with several primary amines in ethanol afforded two kinds of derivatives; monoamination products (compounds 22a- 27a) and symmetric diamination ones (compounds 22b- 26b and 28b). The diamination products were separated easily from the monoamination products, in each reaction, by silica gel chromatography (eluting with chloroform- methanol mixtures). The diamination products are more polar than the monoamination product and the starting material.

As seen in Table 2 the symmetric diamination products are more cytotoxic than the monoamination ones and most of the diamination products are more toxic than their parent compounds 20 and 21 which have IC50’s of 1 ug/ml. Most active are the symmetric derivatives obtained with isobutylamine and methylamine (compounds 22b- 24b) while the more lypophilic derivative obtained with dodecylamine (compound 26a) (as well as 26b) and the more hydrophilic derivative obtained with serinol (compound 28b) are less active.
Table 2. Amination products of compounds 20 and 21 with amines R’NH2 and their in-vitro cytotoxicity against P-388 mouse lymphoma cells.Compound |
R |
R’ |
Yield (%) a b |
IC50 (ug/ml) a b |
||
22 |
H |
(CH3)2CHCH2 |
36 |
38 |
0.25 |
0.1 |
23 |
OCH3 |
(CH3)2CHCH2 |
19 |
37 |
1 |
0.1 |
24 |
H |
CH3 |
46 |
28 |
0.25 |
0.1 |
25 |
H |
CH3OC6H4 |
55 |
38 |
1 |
0.5 |
26 |
H |
CH3(CH2)11 |
24 |
18 |
2.5 |
0.5 |
27 a |
H |
H |
76 |
- |
1 |
- |
28 |
H |
(HOCH2)2CH |
- |
50 |
- |
>10 |
[1] [1a]
T. F. Molinski, Chem.Rev. 1993, 93, 1825-1838.– [1b] D.J. Faulkner, Nat. Prod. Rep. 1999, 155-198, and earlier reports in this series.[2]
G. Gellerman, A. Rudi, Y. Kashman, Tetrahedron Lett. 1993, 34, 1823-1826.[3]
G. Gellerman, M. Babad, Y. Kashman, Tetrahedron Lett. 1993, 34, 1827-1830.[4]
G. Gellerman, A. Rudi, Y. Kashman, Syn. 1994, 239-241.[5]
G. Gellerman, A. Rudi, Y. Kashman, Tetrahedron, 1994, 50, 12959-12972.[6]
J. V. Schurman, E. I. Becker, J. Org. Chem. 1953, 18, 211-217.[7]
Y. T. Pratt, J. Org. Chem. 1962, 27, 3905-3910.[8]
J. R. Peterson, J. K. Zjawiony, S. Liu, C. D. Hufford, A. M. Clark, R. D. Rogers, J. Med. Chem. 1992, 35, 4069-4077.[9]
J. W. Macleod, R. H. Thomson, J. Org. Chem. 1960, 25, 36-42.All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0029 as the message subject of your e-mail.