
Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0035]
Facile and stereoselective synthesis of non-racemic trifluoroalanine
Marcello Crucianelli,a,* Natalia Battista,a Pierfrancesco Bravo,b,c Alessandro Volonterio,b and Matteo Zandab,*
a
Dipartimento di Chimica, Ingegneria Chimica e Materiali, Universita dell'Aquila, Via Vetoio, I-67010 Coppito, ItalyReceived: 25 July 2000 / Uploaded: 29 July 2000
Abstract.
A highly stereoselective enantiodivergent synthesis of non-racemic 3,3,3-trifluoroalanine 7 is reported. The methodology is based on the reduction of the sulfinimine 3 derived from ethyl trifluoropyruvate, followed by acidic hydrolysis of the resulting diastereomeric sulfinamides 4 and 5.
Introduction.
The rapidly expanding interest in the field of peptidomimetics is prompting organic chemists to develop novel and efficient stereocontrolled approaches to rare and unnatural amino acids. An extremely intriguing class of unnatural amino acids is represented by those incorporating one or more fluorine atoms [1]. This interest stems from the well known peculiar biomedicinal and pharmaceutical properties of fluorinated substrates [2], as well as from the considerable synthetic challenges connected with the preparation of these molecules in stereodefined, non-racemic form [3]. In particular, 3,3,3-trifluoroalanine 7 (Scheme 2) and its derivatives have attracted a remarkable deal of interest as suicide inhibitors of a number of pyridoxal-phosphate dependent enzymes [4]. Incorporation of 7 into small peptides has been also achieved, and some of the resulting oligomers have been found to possess interesting biological activity [5]. Although several preparations of racemic 7 have been described following the seminal work of Steglich [6], a very few syntheses of non-racemic 7 are available [7], and only recently its absolute configuration has been clarified [8]. Furthermore, there is currently no practical and straightforward method for the synthesis of non-racemic 7 from readily available or commercial starting materials, such as trifluoropyruvic esters [9]. In this communication we describe the successful accomplishment of such goal starting from ethyl trifluoropyruvate.
Results and discussion.
The chiral Staudinger reagent (S)-2 (e.e. > 95%) [10] (Scheme 1) was synthesized by reaction of the Davis sulfinamide (S)-1 [11] with DEAD/PPh3 (92%) [12]. Then, a high yielding Staudinger (aza-Wittig) reaction of (S)-2 with ethyl trifluoropyruvate was performed in benzene freshly distilled from Na (ca. 90 min. at 40 �C), providing (S)-3. After evaporation of the solvent, the crude reaction mixture containing the highly electrophilic sulfinimine (S)-3 was treated with a variety of reducing agents (Table 1).
Key: i) PPh3, DEAD (92%); ii) CF3COCOOEt, C6H6 freshly distilled from Na, 40 �C (
-Ph3PO).Table 1. Reduction of sulfinimine (S)-3.
Entry |
[H] |
Conditions |
Yield (%)a |
D.r. 4/5 |
1 |
DIBAH |
THF, -70 �C |
52 |
4:1 |
2 |
9-BBN |
THF, 0 �C |
78 |
1:20 |
3 |
DIBAH/ZnBr2 |
THF, r.t. to �70 �C |
58 |
2:1 |
4 |
NaBH4 |
Methanol, -70 �C |
/b,c |
/b,c |
a
Yields from (S)-2. b We recovered 66% of 6 with 33% d.e. c Very low yields in THF, 0 �C to r.t.The most interesting results were achieved with DIBAH (entry 1) and 9-BBN [13] (entry 2) which produced with stereodivergent outcomes the diastereomeric sulfinamides 4 and 5, respectively. In the latter case (9-BBN), the reduction occurred with excellent stereoselectivity (20:1) and overall yield (78%). Although DIBAH provided lower stereocontrol (4:1) and yield (52%), the fact that both diastereomers 4 and 5 are readily accessible from the same enantiomeric sulfinimine (S)-3 makes this method remarkably attractive. DIBAH reduction occurred with lower stereocontrol in favour of 4 (2:1) upon pre-complexation of (S)-3 with ZnBr2. Complex mixtures were obtained with K- and L-Selectride� and LiAlH4 in THF as reducing agents. An undesired side-reaction took place upon treatment of (S)-3 with NaBH4 in methanol (entry 4), namely the addition of methanol across the C=N bond. Thus, a diastereomeric mixture of adducts 6 (Scheme 1) was obtained, whereas the reduction products 4,5 could be neither isolated nor detected [14].
A reasonable transition state (TS) 8 (Scheme 2) accounting for the high stereoselectivity observed with 9-BBN is based on the fact that the sulfinimine (S)-3 is geometrically homogeneous and thermodynamically stable with the sulfinyl and the bulky trifluoromethyl group in trans position with respect to the C=N bond, as demonstrated by theoretical ab initio calculations supported by NMR spectroscopy [15]. In line with the previously proposed TS for highly stereoselective 9-BBN reductions of ketone derived sulfinimines [16], the boron atom should coordinate the sulfinyl oxygen giving rise to a chair-like TS. As a consequence, the hydride predominantly attacks the Re face of the C=N bond producing the diastereomeric sulfinamide 5 with overwhelming preference.
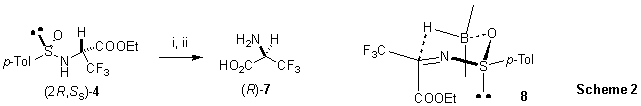
Key: i) HCl conc., reflux, overnight. ii) Dowex 50W-X8.
With the diastereomeric sulfinamides 4 and 5 in hand, both enantiomers of 3,3,3-trifluoroalanine 7 are easily accessible, as demonstrated in the preparation of (R)-7 from 4 (obtained by reduction of 3 with DIBAH), which was treated with 10% HCl (reflux, overnight) [6d] followed by ion exchange chromatography with DOWEX-50W. We were also able to prepare (R)-7 in one-pot in 38% overall yield from (2R,RS)-5 (obtained from the enantiomeric iminophosphorane (R)-2), by submitting the crude 9-BBN reduction mixture to hydrolysis with 10% HCl, followed by the usual ion-exchange purification [17,18]. The stereochemistry of (R)-7 was assessed by comparison of its optical power with the literature values for non-racemic 3,3,3-trifluoroalanine [7,8].
In summary, we have developed an extremely facile, straightforward, stereoselective and enantiodivergent approach to non-racemic 3,3,3-trifluoroalanine, which is now readily available for further biochemical studies and incorporation into peptidomimetics.
Acknowledgment. We thank M.U.R.S.T. COFIN 1998 (Nuove metodologie e strategie di sintesi di composti di interesse biologico) and C.N.R. - C.S.S.O.N. for financial support.
References and notes.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0035 as the message subject of your e-mail.