Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0036]
Synthesis of Enantiomerically Pure Pipecolic Acid Derivatives via Combination of Biocatalysis and Transition Metal Catalysis
Sape S. Kinderman,a Jetze W. van Beijma,a Larissa B. Wolf,a Hans E. Schoemaker,b Henk Hiemstraa and Floris P. J. T. Rutjesa,c*



aInstitute of Molecular
Chemistry,University of Amsterdam Nieuwe Achtergracht
129, 1018 WS Amsterdam, The Netherlands
bDSM Research, Life Science Products P.O.
Box 18, 6160 MD Geleen, The Netherlands
cDepartment of Organic Chemistry, University of Nijmegen Toernooiveld 1, 6525 ED Nijmegen, The Netherlands
E-mail: [email protected]
Received: 26 July 2000 / Uploaded: 30 July 2000
Introduction
The combination of biocatalysis and transition metal catalysis provides a powerful and versatile pathway for the synthesis of multifunctional building blocks. In this approach, the biocatalyst is used to generate enantiomerically pure starting materials, while transition metals are used in a catalytic fashion for further synthetic elaboration.
Biocatalytic Production of Non-Proteinogenic Amino Acids
Racemic amino acid amides can be conveniently resolved into the corresponding (S)-amino
acids and (R)-amino acid amides by an enantioselective aminopeptidase produced by Pseudomonas
putida ATCC 12633. The enzyme is highly selective for a wide range of R-groups and
virtually independent of functionalities in this side chain (Scheme 1).

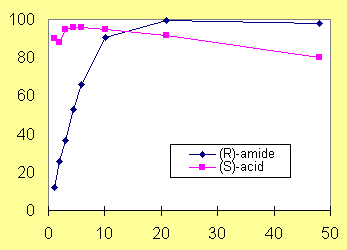
However, the enzymatic resolution gave anomalous results for methionine resembling amino acids due to the presence of a methionine racemase in the whole cells produced by the Pseudomonas putida strain (Fig. 1, exemplified for homopropargylglycine (HPG)). Therefore, a recombinant enzyme system was developed, which appeared a reliable alternative leading to both (S)-amino acids and (R)-amino amides in >99% ee (Fig. 2, shown for HPG).1 Fig. 1: Resolution of HPG with Pseudomonas putida Fig. 2: Resolution of HPG with a recombinant organism

A Novel Route to Modified Pipecolic Acids
Recently,
we developed a novel route for the synthesis of 2,6-disubstituted dihydropyran systems
such as 6.2 A key step in this pathway was the novel Pd-catalyzed
formation of allylic acetals using an alkoxyallene as one of the reaction partners (Scheme
2). Further elaboration via the cyclic acetals 5 via the corresponding oxycarbenium
ion provided the target compounds 6. The mechanism presumably proceeds as indicated
in the same Scheme. Electrophilic activation of the most electron rich double bond (viz.
7), followed by attack of the alcohol and subsequent protonolysis of the resulting
vinylpalladium species 8 gives the acetal 9.

Pipecolic acid (hexahydropyridine-2-carboxylic acid) derivatives are used to introduce conformational restriction in peptides, but may also be used as a starting point for the construction of (libraries of) biologically active compounds and/or natural products. Inspired by the efficient Pd-catalyzed formation of acetals starting from alcohols and by previous RCM-mediated cyclization reactions of allylglycine derivatives,3 we set out to explore a similar route to form the corresponding N,O-acetals, which – if an enantiopure allylglycine deivative (viz. 10, Scheme 3) is used as the starting material - eventually should give rise to unsaturated 2,6-disubstituted pipecolic acid derivatives. Initial results with methoxy-allene were unsatisfactory, leading to the undesired regioisomer 11. However, by using benzyloxy-allene the reaction proceeded smoothly at room temperature to provide the linear allylic N,O-acetal 12. Subjection to standard RCM-conditions then gave the cyclic counterparts 13.

Treatment of the cyclic N,O-acetals with al Lewis acid in the presence of several nucleophiles led to the corresponding functionalized pipecolic acid derivatives 15-19. In general, a highly diastereoselective CC-bond forming reaction took place via the intermediate N-sulfonyliminium ion 14, although in some cases a small amount of the regioisomeric 1,4-adduct was formed.

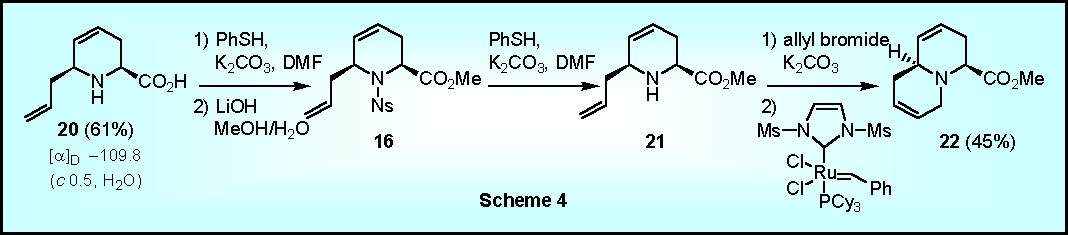
Deprotection of the protected pipecolic acid 16 proceeded readily via PhSH-mediated cleavage of the sulfonamide bond, followed by hydrolysis of the ester to give the amino acid 20 (Scheme 4). Alternatively, desulfonylation followed by allylation and subsequent ring closure via RCM gave the bicyclic amino acid derivative 22 in 45% yield. The latter sequence has not been optimized yet and is currently under investigation.
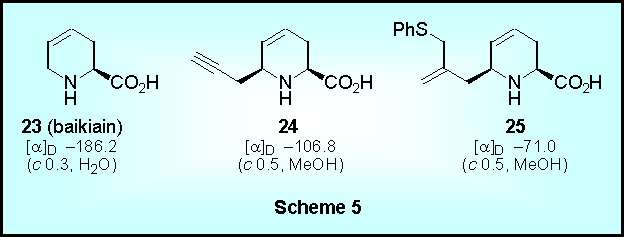
In a similar fashion, deprotection of additional cyclization products led to the enantiomerically pure pipecolic acid derivatives 23-25 including the naturally occurring amino acid baikiain (Scheme 5).
Concluding Remarks
We have developed an efficient enantiomerically pure synthesis of 2,6-disubstituted unsaturated pipecolic acid derivatives, consisting of several transition metal-mediated transformations. At present, we are exploring the possibilities to use these building blocks as starting point for natural product synthesis and the generation of libraries of potentially biologically active compounds.
Acknowledgements
DSM is gratefully acknowledged for providing a research grant to L. B. Wolf. This research has been financially supported by the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (CW-NWO).
References
1. T Sonke, B Kaptein, WHJ Boesten, QB Broxterman, J Kamphuis, F Formaggio, C Toniolo, FPJT Rutjes, HE Schoemaker in Stereoselective Biocatalysis, RN Patel, Ed, Marcel Dekker: New York, 2000, pp. 23-58.
2.FPJT Rutjes, TM Kooistra, H Hiemstra, HE Schoemaker, Synlett 1998, 192.
3.FPJT Rutjes, HE Schoemaker Tetrahedron Lett. 1997, 38, 677.
4.KCMF Tjen, SS Kinderman, HE Schoemaker, H Hiemstra, FPJT Rutjes Chem. Commun. 2000, 699.
All comments on this poster should be sent by
e-mail to (mailto:[email protected] ona.edu) [email protected]
with A0036 as the message subject of your e-mail.