
[A0039]

New Fluorous Chiral Phosphorous Ligands Based on the Binaphthyl Skeleton
E-mail: [email protected]
Received: 2 August 2000 / Uploaded: 3 August 2000
The preparation of two new chiral binaphthyl phosphorous ligands bearing perfluorinated ponytails is reported. Special precautions or harsh reaction conditions were not required and the fluorous ligands were readily isolated from by-products.
The use of non-conventional reaction media such as supercritical fluids [1] and liquid biphase systems (aqueous/organic [3] as well as purely organic [4]) in synthetic organic chemistry is arousing increasing interest.[5] Besides offering the possibility of cleaner technology for the chemical industry, these novel reaction media could also promote reactions that are not attainable in common organic solvents along with selectivity improvements related to the unique solvation environment.[6] The advantages brought about by the use of CO2 (supercritical or compressed) and fluorous biphasic systems (FBS) in catalytic reactions are well-documented. [1] [7] In both instances, a major requirement for the catalyst is the good solubility in the peculiar reaction medium. To this end, several ligands featuring long-chain perfluoroalkyl substituents ("fluorous ligands") have been recently synthesized,[8] including a few examples of chiral compounds.[9] Indeed, the presence of long-chain perfluoroalkyl substituents increases the affinity of an organometallic compound both for CO2 and perfluorocarbons.
As phosphorous-based ligands are extensively used in catalytic chemistry, many efforts has been devoted to the synthesis of their fluorous analogues.[7] [10] However, chiral compounds of this kind are not yet easily available.[11], [12]
Here we describe the synthesis of two new fluorous chiral ligands, namely (R)-2-{bis[4-(1H,1H-perfluorooctyloxy)phenyl]phosphino} -2’-(1H,1H-perfluorooctyloxy)-1,1’-binaphthyl (1) and (R)-2,2’-{ bis[4-(1H,1H-perfluorooctyloxy)phenyl]phosphino} -1,1’-binaphthyl (2) (Figure 1), that enlarge this restricted family of compounds.

The chiral binaphthyl skeleton is the basic structure of many effective enantioselective homogeneous catalysts, as witnessed by the popularity of ligands such as 2-(diphenylphosphino)-2’-alkoxy-1,1’-binaphthyls (MOPs) and 2,2’-bis(diphenylphosphino)-1,1’-binaphthyl (BINAP). [13] [14]. Pd-complexes of chiral MOPs are able to catalyze many types of asymmetric transformations, including regio- and enantioselective alkylation of allylic esters, reduction of the same substrates in the presence of formic acid, hydrosilylation of 1-alkenes, [13c] while BINAP-metal complexes show a broad applicability in various asymmetric reactions such as hydrogenation, transfer hydrogenation, hydroboration and C-C bond formations both in lab scale and in industrial applications. [15] Several applications of related fluorous compounds could be thus envisaged in FBS or scCO2.
Pd-catalyzed coupling of bis(aryl) phosphonic acids with commercially available (R)- or (S)-1,1’-bi-2-naphtol bis(trifluoromethanesulfonate) provides also an easy access to optically pure 2-(diphenylphosphino)-2’-alkoxy-1,1’-binaphthyls [13] and to 2,2’-bis(diarylphosphino)-1,1’-binaphthyls.[16] Leitner and Francio' took advantage of this versatile reaction in the synthesis of a chiral phosphine/phosphite ligand bearing two –(CH2)2C6F13 ponytails, structurally similar to (R,S)-BINAPHOS.[12a] The introduction of the perfluoroalkyl substituents required the use of a properly functionalized bis(aryl) phosphonic acid at an early stage of the synthesis.
In order to increase the flexibility of this approach, we decided to postpone the introduction of perfluorinated residues. Perfluorocarbon-soluble triarylphosphines can be conveniently obtained through O-alkylation of tris(hydroxyphenyl)phosphines. [10d] This procedure allows the insertion of the required perfluoroalkyl chains onto a preformed ligand structure increasing the number of possible locations.
The preparation of optically pure (R)-MOPF15 (1) was thus successfully accomplished by a four steps procedure. The use of bis(4-methoxyphenyl)phosphonic acid 4, which was obtained in a one-pot synthesis from the corresponding para-substituted aryl bromide 3 (Scheme 1),[17] allowed the easy monophosphinylation of bis(trifluoromethanesulfonate) (R)-5 (Scheme 2). This reaction was carried out in the presence of 5 mol % of Pd(OAc)2 and 5 mol % of 1,4-bis(diphenylphosphino)butane (dppb) in dimethylsulfoxide at 100�C. Demethoxylation of the phosphinyl derivative (R)-6 with BBr3 in CH2Cl2 afforded the dihydroxy derivative (R)-7 in 99% yield, after recrystallization from diethyl ether. Subsequent hydrolysis of the remaining triflate group with aqueous sodium hydroxide in a methanol/dioxane mixture converted (R)-7 into (R)-(+)-2-[bis(4-hydroxyphenyl)phosphinyl]-2’-hydroxy-1,1’-binaphthyl (8) in 91% yield. The perfluoroalkoxy chains were then introduced at the 2’ and para positions of phenyl groups by reaction of the three hydroxy groups with perfluoro butansulfonate 9 in the presence of cesium carbonate giving the phosphinyl perfluoroalkoxy (R)-10 in 70% yield after simple flash cromatography (eluent Et2O). Finally, reduction of the P=O functionality was achieved by treatment with Cl3SiH in boiling toluene for 3h, affording pure (R)-1 in 90% yield.


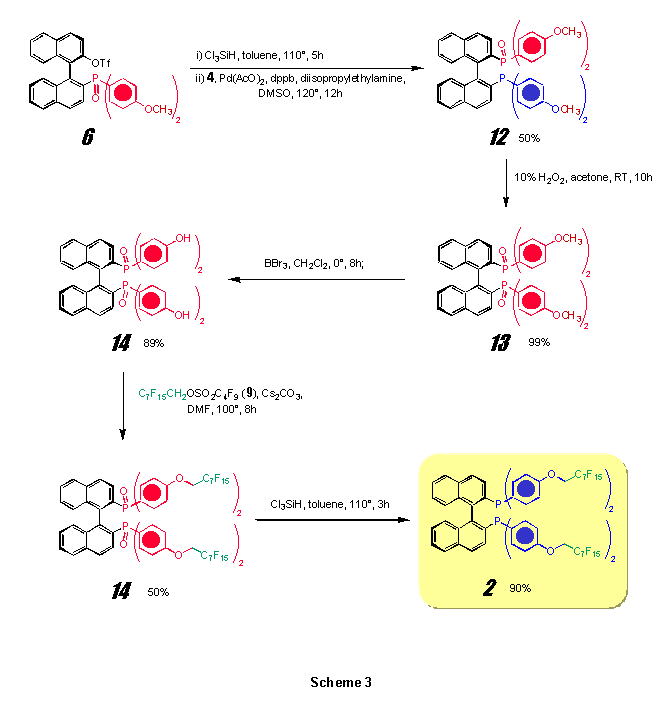
The bidentate fluorous phophine (R)-BINAPF15 (2) was obtained through a synthetic route (Scheme 3) based on that proposed by Gladiali and coworkers for the preparation of 2-diphenilphosphino-2’-diphenylphosphinyl-1,1’-binaphthalene (BINAPO).[16]
The phosphinyl derivative (R)-6, obtained as described above, was converted into the diarylphosphino triflate (R)-11 by reduction with Cl3SiH in boiling toluene. This allows a further Pd-catalyzed coupling reaction with bis(4-methoxyphenyl)phosphonic acid 4 affording the diarylphosphino-diarylphosphinyl compound (R)-12 in 50% overall yield. (R)-12 underwent a quantitative oxidation with H2O2 in acetone to the corresponding (R)-2,2’-[bis(4-methoxyphenyl)phosphinyl]-1,1’-binaphthyl (13). After deprotection of the CH3O- protecting groups, four perfluoroalkoxy substituents were introduced according to the same procedure described for (R)-10. Finally, reduction with Cl3SiH permits a simple access to (R)-BINAPF15 (2).
Possible applications of these new fluorous ligands in fluorous biphasic systems and in supercritical CO2 are currently under investigation.
[1] Chemical Synthesis in Supercritical Fluids (Eds.: Jessop, P. G.; Leitner, W.), Wiley-VCH, New York, 1999.
[2] Welton, T. Chem. Rev. 1999, 99, 2071-2083, and references therein.
[3] a) Jo�, F.; Kath�, A. J. Mol. Catal. A: Chem. 1997, 116, 3-26; b) Cornils, B. J. Mol. Catal. A: Chem. 1999, 143, 1-10, and references therein.
[4] da Rosa, R G.; Martinelli, L.; da Silva, L. H. M.; Loh, W. Chem. Commun. 2000, 33-34, and references therein.
[5] Topics in Current Chemistry, Vol. 206 Modern Solvents in Organic Synthesis (Ed. : Knochel, P.), Springer-Verlag, Berlin, 1999.
[6] See for instance a) Oakes, R. S.; Clifford, A. A.; Bartle, K. D.; Pett, M. T; Rayner, C. M. Chem. Commun. 1999, 247-248; b) de Bellefon, C.; Pollet, E.; Grenouillet, P. J. Mol. Catal. A: Chem. 1999, 145, 121-126.
[7] (a) Horv�th, I. T.; R�bai, J. Science 1994, 266, 72-75. (b) Horv�th, I. T. Acc. Chem. Res. 1998, 31, 641-650.
[8] For recent reviews see: (a) Barthel-Rosa, L. P.; Gladysz, J. A. Coord. Chem. Rev. 1999, 192, 28, 587-605. (b) Fish, R. H. Chem. Eur. J. 1999, 5, 1677-1680. (c) De Wolf, E.; van Koten, G.; Deelman, B.-J. Chem. Soc. Rev. 1999, 28, 37-41. (d) Hope, E. G.; Stuart, A. M. J. Fluorine Chem. 1999, 100, 75-83; (e) Cavazzini, M.; Montanari, F.; Pozzi, G.; Quici, S. J. Fluorine Chem. 1999, 94, 183-193.
[9] (a) Pozzi, G.; Cinato, F.; Montanari, F.; Quici, S. Chem. Commun. 1998, 877-878. (b) Takeuchi, Y.; Nakamura, Y.; Ohgo, Y.; Curran, D. P. Tetrahedron Lett. 1998, 39, 8691-8694. (c) Pozzi, G.; Cavazzini, M.; Cinato, F.; Montanari, F.; Quici, S. Eur. J. Org. Chem. 1999, 1947-1955. (d) Kleijn, H.; Rijnberg, E.; Jastrzebski, J. T. B. H.; van Koten, G. Org. Lett. 1999, 1, 853-855. (e) Takeuchi, Y.; Nakamura, Y.; Ohgo, Y.; Curran, D. P. Tetrahedron Lett. 2000, 41, 57-60. (f) Takeuchi, Y.; Nakamura, Y.; Ohgo, Y.; Curran, D. P. Tetrahedron 2000, 56, 351-356.
[10] Selected examples: (a) Battacharyya, P.; Gudmunsen, D.; Hope, E. G.; Kemmitt, R. D. W.; Paige, D. R.; Stuart, A. M. J. Chem. Soc., Perkin Trans. 1 1997, 3609-3612. (b) Horv�th I.T.; Kiss, G.; Cook, R. A.; Bond, J. E.; Stevens, P. A.; R�bai J.; Mozeleski, E. J. J. Am. Chem. Soc. 1998, 120, 3133-3143. (c) Alvey, L. J.; Rutherford, D.; Juliette, J. J. J.; Gladysz, J. A. J. Org. Chem. 1998, 63, 6302-6308. (d) Sinou, D.; Pozzi, G.;Hope, E. G.; Stuart A. M. Tetrahedron Lett. 1999, 40, 849-852. (e) Richter, B.; Spek A. L.; van Koten, G.; Deelman B.-J. J. Am. Chem. Soc. 2000, 122, 3945-3951. (f) Bergbreiter, D. E.; Franchina, J. G.; Case, B. L. Org. Lett. 2000, 2, 393-395.
[11] Klose, A.; Gladysz J. A. Tetrahedron Asymm., 1999, 10, 2665-2674.
[12] (a) Francio', G.; Leitner, W. Chem. Commun. 1999, 1663-1664. (b) Kainz, S.; Brinkmann, A.; Leitner, W.; Pfaltz, A. J. Am. Chem. Soc. 1999, 121, 6421-6429.
[13] (a) Uozomi, Y.; Tanahashi, A.; Lee, S.-Y.; Hayashi, T. J. Org. Chem. 1993, 58, 1945-1948. (b) Uozomi, Y.; Suzuki, N.; Ogiwara, A.; Hayashi, Y. Tetrahedron, 1994, 50, 4293-4302. (c) Hayashi, T. J. Organomet. Chem. 1999, 576, 195-202 and references therein.
[14] (a) Noyori, R.; Souchi, T.; Takaya, H.; Miyashita, A. Tetrahedron, 1984, 40, 1245-1253; b) Takaya, H.; Mashima, K.; Koyano, K.; Yagi, M.; Kumobayashi, H.; Taketomi, T.; Akutagawa, S.; Noyori, R. J. Org. Chem., 1986, 51, 629-635; c) Noyori, R.; Takaya, H. Acc. Chem. Res., 1990, 23, 345-350 and references therein.
[15] Kumobayashi, H. Recl. Trav. Chim. Pays-Bas, 1996, 115, 201-210 and references therein.
[16] Gladiali, S.; Pulacchini, S.; Fabbri, D.; Manassero, M.; Sansoni, M. Tetrahedron Asymm., 1998, 9, 391-395.
[17] Mann, F.G.; Chaplin, E. J. J. Chem. Soc., 1937, 527-535.
All comments on this poster should be sent by e-mail to (mailto:[email protected]) [email protected] with A0039 as the message subject of your e-mail.