Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0056]
Neighboring Group Participation in the Hydrolysis of Dichloromethyl Carbinols.An Improved Synthesis of Alpha- Hydroxy Aldehydes
George A. Kraus* and Xuemei Wang
Department of Chemistry, Iowa State University, Ames, IA 50011
E-mail: [email protected]
Received: 1 August
2000 / Uploaded: 2 August 2000
Summary: The addition of dihalomethyl lithium reagents to ketones followed by assisted hydrolysis provides a- hydroxy aldehydes in good yield.
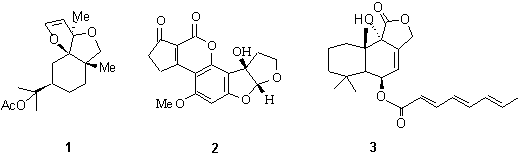
The a- hydroxy aldehyde moiety or its related hemiacetal is contained in a wide variety of natural products including carbohydrates and natural products such as phytuberin (1), aflatoxin M2 (2) and RES-1149-2 (3).1-3 Additionally, this moiety can serve as a building block for the construction of other molecules.
The a- hydroxy aldehyde moiety can readily be synthesized from the reaction of an acyl carbanion equivalent with a carbonyl compound followed by deprotection. The most commonly used acyl carbanion equivalent is the dithiane. Although the original Corey procedure is still used to generate the dithiane anion,4 several procedures have been reported to achieve the hydrolysis5 of the hydroxy dithianes. The diverse array of deprotection conditions arose, in part, because some hydroxy dithianes are unstable to the oxidation or methylation reaction conditions commonly used to deprotect the dithiane.
Recently, Taguchi and coworkers developed a practical and versatile method for introducing a dihalomethyl unit into a carbonyl compound.6 The Taguchi method involves the addition of a strong base such as lithium dicyclohexylamide to a carbonyl compound and methylene chloride or methylene bromide. The resulting alkoxide is unstable and is quenched at low temperature. Using his conditions dihalomethyl carbinols can be generated in 70-90% yields.
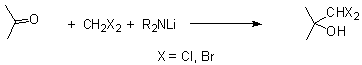
The dihalomethyl carbinols have been employed in ring expansion reactions of cyclic ketones, in the synthesis of 1,1-dichloroalkenes and in the generation of a- hydroxy aldehydes. The latter application has been little used because by-products such as a- halo aldehydes and a,b- unsaturated aldehydes are co-produced in comparable yields.7 Under basic conditions a dichloromethyl carbinol is converted into a chloroepoxide which then gives rise to both the a- hydroxy aldehyde and the a- chloro aldehyde. We reasoned that a proximate acohol might intramolecularly intercept the chloroepoxide intermediate.
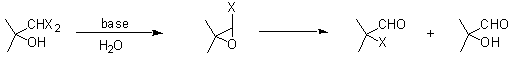
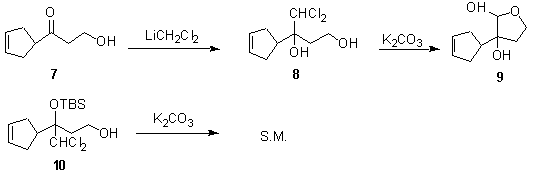
Initially, we prepared diol 4 from the reaction of dibromomethyl lithium and enone 5.8 Hydrolysis using potassium carbonate in aqueous isopropanol afforded hydroxy hemiacetal 6 in 93% isolated yield. By comparison, the yield of hydroxy aldehyde from the adduct of dibromomethyl lithium with cyclohexanone was 40%. The production of 6 represented model system experiments toward a synthesis of 3.

We later found that the dihalomethyl lithium addition reaction proceeded more cleanly and in higher yield when dichloromethyl lithium was employed. When ketone 7 was treated with dichloromethyl lithium, diol 8 was generated in 73% yield. Hydrolysis of 8 with potassium carbonate in aqueous isopropanol afforded hemiacetal 9 in 78% yield. Interestingly. When 10 was treated with potassium carbonate, only starting material was recovered, even when the reaction was warmed to 60 °C. This experiment reinforces the idea that a chloroepoxide intermediate is involved in the hydrolysis reaction.
Diol 12 was then prepared from ortho-hydroxyacetophenone (11) in 93% yield. Hydrolysis of diol 12 afforded hemiacetal 13 in 70% yield, as a mixture of stereoisomers. The related triol 14 was synthesized in 73% yield from diol 15. Hydrolysis of 14 afforded 16 in 93% yield.
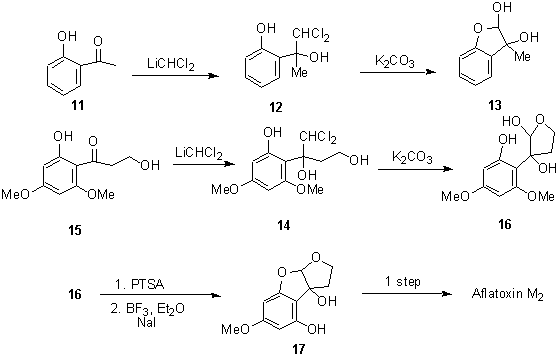
Compound 16 is an intermediate for the preparation of aflatoxin M2.9 When 16 was treated with p-toluenesulfonic acid in warm methylene chloride, and then demethylated using boron trifluoride etherate and NaI, phenol 17 was isolated in 55% yield from 16. The structure of 17 was supported by 2D NOESY experiments, showing that the desired demethylation had occurred. Buchi and coworkers have converted compound 17 into aflatoxin M2 in one step.10
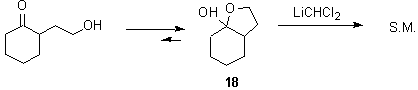
Hydroxy ketone 18 exists almost exclusively as the hemiketal. It was of interest to determine whether the unstable dichloromethyl lithium reagent would add to this compound. Unfortunately, the reaction of dichloromethyl lithium with 18 returned recovered starting material.
A carboxylic acid was also evaluated as a proximate group in the hydrolysis reaction. Compound 19 was prepared in 91% yield from the reaction of five equivalents of dichloromethyl lithium with commercially available ortho-acetyl benzoic acid (20). Hydrolysis of 19 with potassium carbonate in aqueous isopropanol provided acid 21 in 72% yield. In a related set of experiments, levulinic acid was treated with five equivalents of dichloromethyl lithium. The resulting hydroxy acid was 22 hydrolyzed to afford acid 23 in good yield.

In summary, the sequence of dihalomethyl lithium addition followed by assisted hydrolysis constitutes an effective method for the synthesis of a- hydroxy aldehydes. Both alcohols and carboxylic acids can assist the hydrolysis. This method should be compatible with a wide variety of substrates.
Experimental
General procedure for addition of dihalomethyl lithium to carbonyl compounds: To a solution of carbonyl compound (10 mmol) and dihalomethane (50 mmol) in THF (50 mL), 3 to 5 equivalents of LiTMP [30 mmol, prepared from 2,2,6,6-tetramethylpiperidine (4.23 g, 30 mmol) and n-butyllithium (12 mL of 2.5 M solution in hexanes)], were added slowly at –78 oC. The reaction mixture was stirred at –78oC for 3 h and monitored by TLC. After the reaction was complete, saturated ammonium chloride solution (15 mL) was added at –78 oC. The reaction was then extracted with diethyl ether, washed with brine, dried over anhydrous magnesium sulfate, and concentrated in vacuo. The residue was purified by sgc, eluting with a mixture of hexanes and ethyl acetate, to give the pure dihalomethyl carbinol.
1-(Dibromomethyl)-2-(hydroxymethyl) cyclohex-2-enol (4): 1H NMR (CDCl3) 6.10 (s, 1H), 5.95 (d, 1H), 4.30 (dd, J = 1, 11.2 Hz, 2H), 4.17 (s, 1H), 1.5-2.53 (m, 4H). 13C NMR (CDCl3,) 216.2, 146.9, 111.0, 68.4, 50.1, 44.5, 43.5, 33.2, 25.0, 21.2, 20.6.
1-(Dichloromethyl)-1-(3-cyclopentenyl)propane-1,3-diol (8): 1H NMR (CDCl3,) 5.80 (s, 1H), 5.69 (s, 2H), 4.15 (m, 1H), 3.97 (s, 1H), 3.77 (br s, 1H), 2.85 (p, J = 6.4 Hz, 1H), 2.49 (m, 4H), 2.3 (m, 2H), 2.0 (m, 2H). 13C NMR (CDCl3,) 129.7, 129.5, 79.7, 78.8, 59.7, 45.3, 41.6, 34.7, 32.8.
1-(Dichloromethyl)-1-(1-hydroxyphenyl) ethanol (12): 1H NMR (CDCl3) 8.19 (br s, 1H), 7.1-7.2 (m, 2H), 6.8-6.9 (m, 2H), 6.13 (s, 1H), 3.54 (br s, 1H), 1.87 (s, 3H). 13C NMR (CDCl3,) 154.6, 130.3, 128.4, 125.6, 120.6, 117.7, 81.0, 78.8, 23.1. IR (neat) cm-1: 3318, 3201, 1584, 1492, 1456, 1231, 789, 751. MS m/z (CI-NH3): 220, 167, 137, 119. HRMS m/z M+: 220.0056, calcd. for C9H10O2Cl2: 220.0058.
General procedure for the hydrolysis of dihalomethyl carbinols:
To the dihalomethyl carbinol (100 mg) in a mixture of isopropanol (5 mL) and water (5 mL), potassium carbonate (100 mg) was added and then stirred at rt from 1 to 12 h. After the reaction was complete (TLC), isopropanol was removed in vacuo. The aqueous solution was saturated with sodium chloride and extracted with ethyl acetate. The combined organic layers were dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was purified by sgc (eluting with a mixture of hexanes and ethyl acetate) to give a pure compound.
1,7a-Dihydroxy-1, 3, 5, 6, 7, 7a-hexahydroisobenzofuran (6): 1H NMR (CDCl3) 4.80 (s, 1H), 4.65 (s, 1H), 3.64 (d, J = 11.2 Hz, 1H), 3.47 (d, J = 10.8 Hz, 1H), 2.50 (m, 2H), 1.6-1.8 (m, 2H), 1.70 (s, 3H), 1.2-1.5 (m, 2H), 1.08 (s, 3H). 13C NMR (CDCl3) 216.2, 146.9, 111.0, 68.4, 50.1, 44.5, 43.5, 33.2, 25.0, 21.2, 20.6.
3-Methyl-2, 3-dihydro-benzofuran-2, 3-diol (13): 1H NMR (CDCl3) 6.05 (d, J = 2.4 Hz, 1H), 6.05 (d, J = 2.4 Hz, 1H), 5.22 (s, 1H), 4.2 (m, 2H), 3.82 (s, 3H), 3.77 (s, 3H), 2.5-2.8 (m, 2H). 13C NMR (CDCl3,) 216.2, 146.9, 111.0, 68.4, 50.1, 44.5, 43.5, 33.2, 25.0, 21.2, 20.6.
3-(2-Hydroxy-4, 6-dimethoxyphenyl)tetrahydrofuran-2, 3-diol (16): 1H NMR (CDCl3,) 6.05 (d, J = 2.4 Hz, 1H), 6.05 (d, J = 2.4 Hz, 1H), 5.22 (s, 1H), 4.2 (m, 2H), 3.82 (s, 3H), 3.77 (s, 3H), 2.5-2.8 (m, 2H). 13C NMR (CDCl3,) 162.2, 146.9, 111.0, 68.4, 50.1, 44.5, 43.5, 33.2, 25.0, 21.2, 20.6.
References
1. Fujimori, T.; Tanaka, H.; Kato, K. Phytochemistry 1983, 22, 1038.
2. Schuda, P. F. Top. Curr. Chem. 1980, 91, 75-111.
3. Hayes, M. A.; Wrigley, S. K.; Chetland, I.; Reynolds, E. E.; Ainsworth, A. M.; Renno, D. V.; Latif, M. A.; Cheng, X.-M.; Hupe, D. J.; Charlton, P.; Doherty, A. M. J. Antibiot. 1996, 49, 505-512.
4. Seebach, D.; Corey, E. J. J. Org. Chem. 1975, 40, 231-7.
5. Degani, I.; Fochi, R.; Regondi, V. Synthesis 1981, 51-3
6. Taguchi, H.; Yamamoto, H.; Nozaki, H. J. Am. Chem. Soc. 1974, 96, 3010-11.
7. Blumbergs, P.; LaMontagne, M. P.; Stevens, J. I. J. Org. Chem. 1972, 37, 1248-51.
8. Smith, A. B., III; Dorsey, B. D.; Ohba, M.; Lupo, A. T., Jr.; Malamas, M. S. J. Org. Chem. 1988, 53, 4314-25.
9. Kraus, G. A.; Wang, X. Tetrahedron Lett., 1999, 40, 8513.
10. Buchi, G.; Weinreb, S. M. J. Am. Chem. Soc. 1969, 91, 5408-9.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0056 as the message subject of your e-mail.