Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0058]
Diels-Alder cycloadditions of 1,3-cyclohexadien-4,5-diones (o-benzoquinones) with norbornadiene. Part II. A high level computational study of their stereospecificities.
Davor Margetic, Martin R. Johnston, Melissa J. Latter and Ronald N. Warrener
Centre for Molecular Architecture, Central Queensland
University, North Rockhampton, 4701, QLD, Australia
E-mail: [email protected]
Received:
1 August 2000 / Uploaded: 2 August 2000
Abstract: Ab initio and DFT quantum chemical calculations have been applied to a study of Diels-Alder reactions of o-benzoquinone as diene and norbornadiene as dienophile. Transition states for these reactions are located and activation energies estimated. The prefered exo-¶- facial selectivities and exo,endo- stereospecificities exhibited in these cycloadditions are readily predicted using RHF/3-21G or higher levels of calculations. Differences between experimentally observed results and calculations may be explained by the postulation of a second, nonconcerted biradical mechanism leading to formation of hetero Diels-Alder products.
Introduction. Diels-Alder methodology has been used by ourselves[1] and others[2] to produce rigid polyalicyclic structures. The overall shape of such systems depends on the specificity of the reactions used in each step of the construction. With a knowledge of stereospecificity, for example, it is possible to introduce a bend in the framework or to extend the structure linearly.
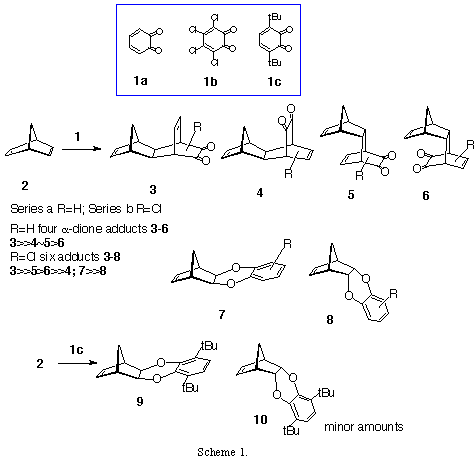
In the previous paper (Part I) we have described synthetic methods for preparation of o-benzoquinone, o-chloranil and 2,5-di-(t-bu)-benzoquinone adducts with norbornadiene.[3,4] Here, we will present the results of the computational study of these reactions using high level quantum mechanical calculations. We have found that o-benzoquinone 1a generated in situ gives four a-dione adducts 3-6 (Scheme 1) with adduct 3 being the dominant product and adducts 4 and 5 are found in smaller amounts and 6 only spectroscopically detected. In contrast to this reaction, o-chloranil 1b gives six adducts 3-8, while 2,5-di-(t-bu)-benzoquinone 1c gave only adducts 9 and in minor amounts 10.
High levels ab initio [5] and B3LYP/6-31G* DFT methods[6] have proved to give excellent results for energy barrier estimation of pericyclic reactions. We have previously used ab initio and DFT calculations to successfully predict stereospecificities of different Diels-Alder reactions of cyclic dienes with cyclic dienophiles.[7]
In order to explain the experimentally observed stereospecificites, transition states for these reactions have been located and activation energies estimated at various levels of theory.
Computational methodology. All ab initio calculations were conducted using the SPARTAN program[8] on a Silicon Graphics Oxygen R5000 workstations. These geometries have been used as the initial geometries for DFT calculations with Gaussian98.[9] MP2 calculations were conducted using IBM SP2 supercomputer. Geometric optimizations were carried out without using symmetry or other structural restrictions. All calculations are performed at the restricted Hartree-Fock level[10] with 3-21G and 6-31G* basis sets[11]. Each transition structure was located using a standard routine within Gaussian 98.[12] For all structures the vibrational analysis was performed with the same basis set used for optimization. The activation energies were also estimated from 6-31G* and MP2/6-31G* single point calculations on the RHF/3-21G and RHF/6-31G* optimized geometries. Further optimizations were carried out with density functional theory (DFT) hybrid B3LYP (Becke's 3 parameter functional[13] with the non-local correlation provided by the expression of Lee et al.)[14]. Single point energy calculations were also estimated using DFT B3LYP method: B3LYP//3-21G[15] and B3LYP/6-31G*. In order to minimize computational efforts, all substituents (chlorine or t-Bu) were omitted and only the parent o-benzoquinone reaction was studied.
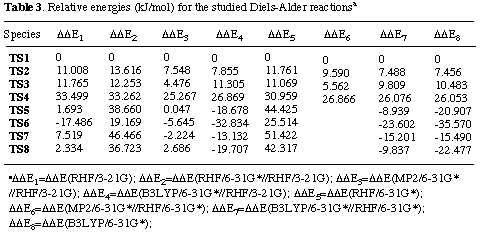
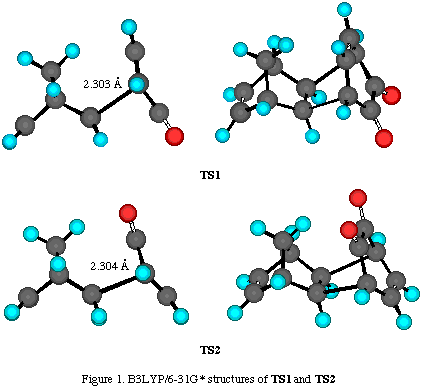
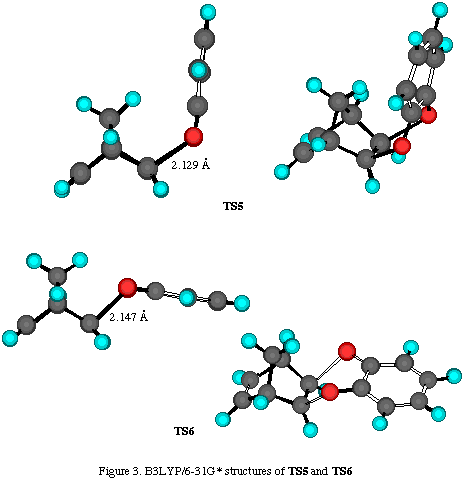
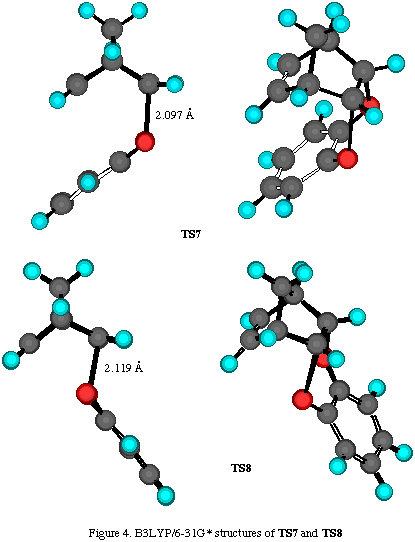
Results and discussion. The total energies of the calculated molecules and their associated transition states (Scheme 2) are collected in Table 1, while activation energies and relative energies are presented in Tables 2 and 3. B3LYP transition state structures are depicted in Figures 1-4. All located transition state structures correspond to the concerted, synchronous mechanism. The lenghts of the bonds undergoing first-order changes in these transition structures are commensurate with those expected for concerted cycloaddition reaction transition states.
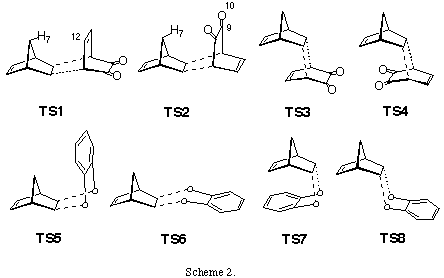
FMO analysis has shown that norbornadiene HOMO and o-benzoquinone LUMO are the most important interacting orbitals, i.e. this represents an inverse electron-demand Diels-Alder reaction (norbornadiene HOMO -6.73 eV, LUMO 0.0 eV, o-benzoquinone HOMO -6.79 eV, LUMO -3.54 eV). Furthermore, quantum of charge transfer (qCT) from diene to dienophile for TS1 - 4 are estimated to be -0.124, -0.108, -0.136 and -0.120 eV, respectievly, clearly indicating inverse mechanism.

¶-facial selectivity. The exclusive exo- side preference in cycloaddition reactions on the norbornene double bond[16] however was not observed in the reactions with the o-benzoquinone presented here. The o-benzoquinone (1a) and o-chloranil (1b) alongside exo,exo- (3) and exo,endo- (4) adducts the reaction mixtures also contained detectable amounts of endo,exo- (5) and endo,endo- (6) adducts.
The analysis of results collected in Tables 1 - 3 shows that all methods employed, starting with RHF/3-21G gave a good prediction of exo- ¶-facial selectivity in norbornene system. Regardless of the computational level employed, exo,exo- adduct 3 (which is formed via TS1) was found to be the prefered by 7.5-13.6 kJ/mol over exo,endo- adduct 4, what was observed experimentally. The largest difference (13.6 kJ/mol) was estimated at the 6-31G*//3-21G level. These results reinforce our previous findings that 6-31G*//3-21G and higher theoretical levels correctly model ¶-facial selectivity in cycloaddtion reactions with norbornenes.[7] As expected, estimated energy barriers vary with the computational level applied.[17] In contrast to these findings, there was found no significant difference was found between activation energies for the formation of exo,endo- 4 and endo,exo- 5 adducts. These predictions are less consistent, depending on the calculation level employed. While 3-21G full otimization, single point B3LYP/6-31G* energy estimations on the 3-21G and 6-31G* optimized structures and B3LYP/6-31G* calculations predict smaller activation energies for TS2 than TS3 (by 0.76, 3.5, 2.3 and 3.03 kJ/mol. respectively), the 6-31G*//3-21G, MP2/6-31G*//3-21G, 6-31G* and MP2/6-31G*//6-31G* calculations (Table 3) predict smaller activation energies for TS3 over TS2 (by 1.4, 3.1, 0.7 and 4.0 kJ/mol, respectively). However, we may conclude that, since TS2 - TS3 energy differences are relatively small, all calculations predict formation of similar amounts of products 4 and 5. Furthermore, the large differences in activation energies between TS1 and TS4 (within a range of 25.3 and 33.5 kJ/mol, as estimated by MP2/6-31G*//3-21G and 3-31G methods, respectively, Table 3), clearly indicate that formation of product 6 is greatly disfavoured. This prediction is in accord with our experimental results as well as computational results obtained by Hehre et al. having recently shown that ab initio calculations using 3-21G model correctly predict the relative energies of the transition states for the two modes of attack to diastereotopic faces of a diene.[18]

The origin of stereospecifity may be rationalized using Mulliken population analysis, which gives a qualitative indicator of the amount of electron density shared by two atoms, and also provides some evidence for secondary orbital interaction between two reactantants. Such an analysis was employed succesfully by Houk et al. to explain the stereoselectivities of several Diels - Alder reactions.[19]
In TS1 the methylene hydrogen - double bond carbon (H7C12) overlap density has a positive value of 0.0014, indicating an attractive interaction, while in TS2 the methylene hydrogen - carbonyl carbon atom (H7C9) overlap population is repulsive with value of -0.003. Furthermore, the methylene carbon - carbonyl carbon (C7C9) overlap population is also repulsive with value of -0.002, and methylene carbon - carbonyl oxygen (C7O10) overlap population is repulsive with value of -0.005, indicating larger destabilizing secondary orbital interactions in TS2. However, the similar analysis of overlap densities employed on the TS3 and TS3 does not give such conclusive differences.
Formation of hetero Diels-Alder products. We have found that in the cycloaddition reactions o-chloranil 1b and 2,5-di-(t-bu)-benzoquinone 1c also serve as heterodienes. While 1b gave a mixture of normal Diels-Alder adducts 3-6, and smaller quantities of exo- and endo- adducts 7 and 8, the quinone 1c gave only hetero Diels-Alder adducts 9 (major product) and 10. Similar behaviour of o-benzoqunione was observed previously by Kumar et al. [20]
For each mode of attack to the ¶-system, we have located two transition structures: for exo- approach TS5 (where aromatic ring is facing methylene bridge) and TS6 (where aromatic ring lay in the plane of norbornene cyclohexene ring), and for the endo- addition TS7 (where aromatic moiety is facing double bond) and TS8 (where aromatic ring is outside norbornene moiety) (Scheme 2). While these structures possess some energy differences, we were unable to see discrete species using 1H-NMR, which suggest their rapid interconversion at room temperature.
The inspection of results collected in Tables 2-4 reveals nonconsistent results for activation energies for TS5-8 at all employed theoretical levels. For most transition structures, activation energies are smaller, or very similar to the one for the TS1-4, which is opposite to experimental results. For instance, the B3LYP/6-31G* method, which gives excellent results for cycloaddition reactions, predicts TS6 to have the smallest energy followed by TS5, TS7 and TS8, while activation energies for TS1-4 are significantly greater, suggesting the exclusive formation of products 3-6. However, these adducts have not been experimentaly detected. In this case, only the RHF/6-31G* method gave the correct predictions, i.e. exclusive formation of products 3-5, while hetero Diels-Alder products have much bigger activation energies.
The lengths of the bonds undergoing first-order changes in these transtition structures are those expected for concerted cycloaddition transition states (Figures 3 and 4). However, harmonic frequency calculations identified these structures as second-order saddle points. Efforts to locate concerted, synchronous transition structures for hetero Diels-Alder reaction using various spin-restricted wave functions resulted in each case, in the location of a single stationary point possessing more than one negative mode of vibration. Despite an extensive search of the singlet-state energy surface using resticted levels of ab initio and DFT theory, we were unable to locate genuine transition structures for either synchronous or a nonsynchronous concerted cycloaddition process. The same finding was experienced using various spin-restricted wave functions (either 3-21G, 6-31G* or B3LYP methods).

The discrepancies between the experimentally observed results and the theoretical analysis may be explained by the postulation of a second, nonconcerted biradical mechanism leading to formation of hetero Diels-Alder products 5-10. Given our failure to locate concerted transition strucutre for the o-benzoquinone hetero Diels-Alder cycloaddition, our attention is now focused on calculations of nonconcerted reactions. The calculations employing unrestricted ab initio and B3LYP calculations are currently being undertaken and these results will be reported in due course.
Conclusion. The present results demonstrate the ability of ab initio calculations to accurately predict relative reactivities and stereospecificities for inverse electron-demand Diels - Alder reactions in alicyclic systems with cyclic 1,3-dienes. Transition states were located and activation barriers estimated at different levels of theory, by Hartree-Fock, post- Hartree-Fock and DFT methods. The high exo-¶-facial selectivity exhibited in these cycloadditions are readily predicted using RHF/3-21G or higher ab initio levels. In the case of hetero Diels-Alder products, all quantum chemical levels employed failed to correctly predict energy barriers, which suggests that second, nonconcerted biradical mechanism may be operating.
Acknowledgements. We are grateful to the Australian Research Council (ARC) for funding.
References.
1. Warrener, R. N.; Butler, D. N., Russell, R. A. Synlett, 1998, 366.
2. Carruthers, W. "Cycloaddition Reactions in Organic Synthesis", Pergamon, Oxford, 1990; Wasserman A. " Diels - Alder Reactions", Elsevier, N. York 1965; Fringuelli, F.; Taticchi, A. "Dienes in the Diels - Alder Reaction", Wiley, N. York, 1990;
3. Johnston, M. R.; Latter, M. J.; Warrener, R. N.; Golic, M.; McKavanagh. D.; Margetic, D. ECSOC-4 preceeding paper, Part I.
4. Warrener, M. R.; Johnston, M. R.; Schultz, A. C.; Golic, M.; Houghton, M.; Gunter, M. J. Synlett 1998, 590; Nunn, E. E.; Wilson, W. S.; Warrener, R. N. Tetrahedron Lett. 1972, 2, 175.
5. Houk., K. N.; Li, Y.; Evansceck, J. D. Angew. Chem. Int. Ed. Engl. 1992, 31, 1, 682.
6. Goldstein, E.; Beno, B.; Houk, K. N.; J. Am. Chem. Soc. 1996, 118, 6036; Houk, K. N.; Beno, B. R.; Nendel, M.; Black, K.; Yoo, H. Y.; Wilsey, S.; Lee, J. K.; J. Mol. Struct. (Theochem) 1997, 398-399, 169; Domingo, L. R.; Arno, M.; Andres, J. J. Am. Chem. Soc. 1998, 120, 1617; Domingo, L. R.; Picher, M. T.; Andres, J. J. Org. Chem. 2000, 65, 3473; Domingo, L. R.; Picher, M. T.; Andres, J. Oliva, M. J. Org. Chem. 1999, 64, 3026; Liu, J.; Niwayama, S.; You, Y.; Houk, K. N. J. Org. Chem. 1998, 63, 1064; Bareone, V.; Arnaud, R.; Chavant, P. Y.; Vallee, Y. J. Org. Chem. 1996, 61, 5121; Konecny, R.; Doren, D. J. J. Am. Chem. Soc. 1997, 119, 11098; Jones, G. A.; Shephard, M. J.; Paddon-Row, M. N.; Beno, B. R.; Houk, K. N.; Redmond, K.; Carpenter, B. K.; J. Am. Chem. Soc. 1999, 121, 4334.
7. Warrener, R. N.; Russell, R. A.; Margetic, D. Synlett 1997, 1, 38; Margetic, D.; Warrener, R. N.; Malpass, J. R. Internet Journal of Computational Chemistry, Bachrach, S., Ed, November 2 - 30, 1998, http://hackberry.chem.niu.edu/; Sun, G.; Butler, D. N.; Warrener, R. N.; Margetic, D.; Malpass, J. R., Article 062, "Electronic Conference on Heterocyclic Chemistry '98 ", Rzepa, H. S. and Kappe, O. (Eds), Imperial College Press, 1998, ISBN-981-02-3549-1; http://www.ch.ic.ac.uk /ectoc/echet98; Margetic, D.; Warrener, R. N.; Tiekink, E. R. T., Article 064, "Electronic Conference on Heterocyclic Chemistry '98 ", Rzepa, H. S. and Kappe, O. (Eds), Imperial College Press, 1998, ISBN-981-02-3549-1; http://www.ch.ic.ac.uk/ectoc/echet98; Margetic, D.; Warrener, R. N.; Malpass, J. R., Fifth Electronic Conference on Computational Chemistry (ECCC5), Bachrach, S., Ed, November 2 - 30, 1998, http://hackberry.chem.niu.edu/;, Golic, M.; Butler, D. N.; Warrener, R. N.; Margetic, D., Third International Electronic Conference on Synthetic Organic Chemistry (ECSOC-3), http://www.reprints.net/ecsoc-3.htm, September 1-30, 1999.
8. Spartan v. 5.0, Wavefunction, Inc. 18401 Von Karman Avenue, Suite 370, Irvine, CA, 92612, 1997.
9. Gaussian 98, Revision A.5, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1998.
10. Roothan, C. C. J. Rev. Mod. Phys. 1951, 23, 69.
11. Hehre, W. J.; Radom, L.; Schleyer, P.v.R.; Pople, J. "Ab Initio Molecular Orbital Theory", Wiley, N. York, 1986.
12. McIver, J. W.; Komornicki, A. J. Am. Chem. Soc. 1972, 94, 2625-2629.
13. Becke, A. D. J. Chem. Phys. 1993, 98, 1372.
14. Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
15. Marchand, A. P.; Ganguly, B.; Shukla, R. Tetrahedron 1998, 54, 4477; Sustmann, R.; Sicking, W. J. Am. Chem. Soc. 1996, 118, 12562.
16. For origins and consequences of norbornene p - facial stereoselectivity see: Borden, W. T. Chem. Rev. 1989, 89, 1095 and references cited therein.
17. Bach, R. D.; McDouall, J. J. W.; Schlegel, H. B. J. Org. Chem. 1989, 54, 2931.
18. Hehre, W. R. "Practical Strategies for Electronic Structure Calculations" Wavefunction, Inc. 18401 Von Karman Suite 370, Irvine, California 92715, 1995.
19. Loncharich, R. J.; Brown, F. K. Houk, K. N. J. Org. Chem. 1989, 54, 1129 - 1134; Birney, D. M.; Houk. K. N. J. Am. Chem. Soc. 1990, 112, 4127 - 4133.
20. Nair, V.; Kumar, S. Tetrahedron 1996, 52, 4029; Nair, V.; Kumar, S. J. Chem. Soc., Chem. Commun. 1994, 1341; Nair, V.; Kumar, S. Synlett 1996, 1143.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0058 as the message subject of your e-mail.