Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0060]
Functionalization of Double Bonds via Cationic Sulfenyl-X Additions.
Alexei Novikov and David Goldsmith*
Department of Chemistry, Emory University, Atlanta GA 30322 USA
* To whom inquires should be addressed: [email protected]
Received: 2
August 2000 / Uploaded: 3 August 2000
Introduction
The addition of cationic sulfenyl halides or other derivatives to double bonds is a well-known reaction [1]. The chemistry of b-heteroatom substituted organic sulfides, the products of these reactions has also been studied [2]. However, relatively little attention has been given to utilizing the addition process as a synthetic method.
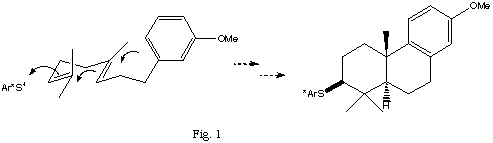
The work reported here was targeted at studying two potential synthetic applications of cationic arylsulfenyl species addition reactions; first, the preparation of chiral epoxides and, second, the enantioselective cationic cyclization, of polyene substrates. . Sulfenyl cations have previously been used by Livinghouse for cyclization processes but not in chiral fashion.[3].
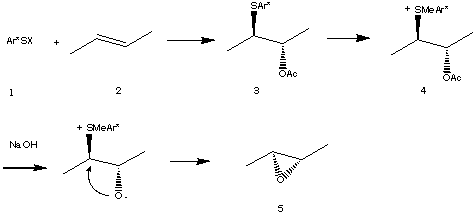
For the preparation of epoxides using arylsulfenyl-heteroatom addition the process would involve initial attack of the sulfenyl halide, 1, on the double bond of an alkene, 2, followed by trapping of the resulting episulfonium ion with an oxygen nucleophile, e.g., acetate. The resulting b-acetoxy-arylsulfide, 3, would then be converted to a sulfonium salt, 4, by alkylation. Treatment of 4 with base to affect elimination would result in the formation of an epoxide, 5. By analogy to recent work with selenenyl reactants enatioselectivity would be expected in this process if chiral arylsulfenyl compounds were to be employed[1].
Cationic cyclizations of polyenes have been studied using a wide variety of initiating species including both protic and Lewis acids, mercuric salts, halogens and arylsulfenyl species. In the latter cases, however, the initiating sulfenyl cation did not contain stereogenic centers and as a consequence no stereoselectivity at the initial reactive carbon of the polyene substrate could be observed [4]. Our object was to investigate the possibility of diastereoselectivity in cyclization as a consequence of using a chiral sulfenyl cationic initiator (Fig. 1).
Preparation of epoxides
The addition of a sulfenyl cation to double bonds was first attempted using the procedure employing phenylsulphenyl chloride, acetic acid and silver tetrafluoroborate described by Smit and co-workers [5]. While it worked well in case of a simple alkene like cyclooctene, 6, (Entry 1, Table 1) lower yields of addition products were obtained in the cases of more complex substrates, even one as simple as the trisubstituted alkene, methylcyclohexene, 7 (Entries 2 and 3, Table 1). In addition, in the attempt to use methodology involving less expensive reagents than silver tetrafluoroborate. a simpler and more effective procedure was developed.
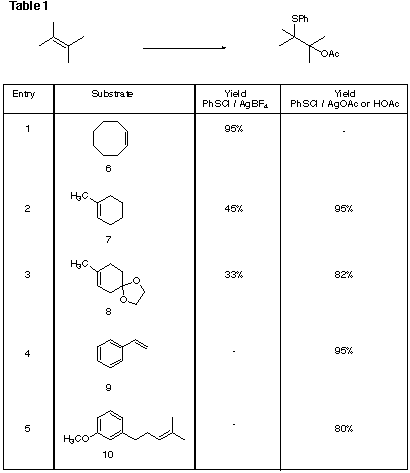
Phenyl sulfenyl chloride was first added to the alkene producing the sulfenyl chloride, followed by solvolysis of the chlorosulfide in acetic acid in the presence of silver or sodium acetate. This procedure produced substantially higher yields for the formation of acetoxy sulfides from 7, 8, 9, and 10 (Entries 2 &emdash; 5, Table 1) in part, we believe, due to the suppression of byproducts resulting from carbocationic intermediates. It was also experimentally easier to carry out. However, in the case of phenylcyclohexene, 11, where a highly stabilized carbocation may be readily formed, alkene 12 was obtained, and with b-pinene, 13, a substrate particularly prone to cationic rearrangement, isomerization was observed, leading to acetoxy sulfide, 14.

Experiments designed to convert the prepared acetoxysulfides to epoxides were then carried out: (Table 2).

Although the reactions were not clean, several sulfonuim salts, 18, 19, and 20, (Entries 1, 2, and 3, Table 2) were successfully prepared from the corresponding acetoxysulfides, 15, 16, and 17. However, treatment of the sulfonium salts with potassium hydroxide gave either low yields or none of the desired epoxides, 21, 22, and 23.
To gain insight into the factors controlling this process we examined sulfonium salt, 20, obtained from 17, the addition product from styrene. Treatment of salt 20 with sodium hydroxide in methanol or sodium methoxide in methylene chloride gave a mixture of unsaturated sulfide 24 and the methoxysulfide 24. With BuLi in THF or potassium carbonate in DMF 20 gave a mixture of the acetoxy sulfide 17 and the same unsaturated sulfide 25. To verify the origin of the methyl in the product, the reaction was carried out in ethanol. The ethoxysulfide 26 was isolated instead of 24 confirming that the methyl group was introduced from methanol, not via intermolecular methylation.
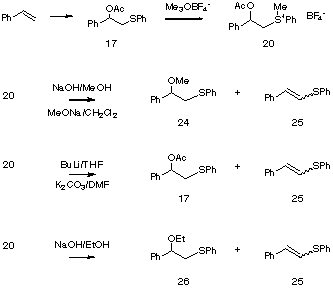
Apparently, under the reaction conditions employed, cleavage of the acetate group of 20 is slow compared to either demethylation to form 17, or elimination/addition to form 24 or 26. Indeed, replacement of the acetate with the more reactive and less hindered formate, 27, or with the unprotected hydroxyl group itself, 28, led to the desired epoxide, 29, upon treatment with base.

Cyclization
The cyclization substrates 30 and 31 were prepared by Li2CuCl4 catalyzed coupling of m-methoxybenzyl magnesium chloride, 32, with the corresponding allylic chlorides 33, and 34 (Scheme 1).
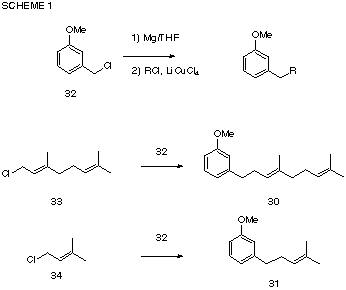
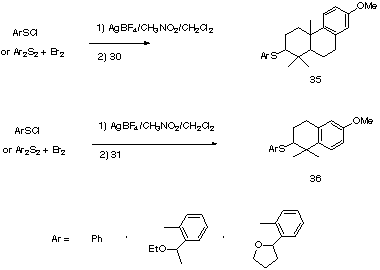
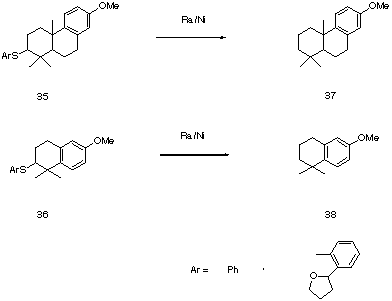
Cyclization, initiated with the phenylsulfenyl cation generated by reaction of phenylsulfenyl chloride with silver tertafluoroborate gave a mixture of products, from which the expected cyclization products 35 and 36 were isolated. The structures were confirmed by hydrogenolysis of the sulfide linkages with Raney Ni and comparison of the resulting hydrocarbons, 37 and 38 with literature data [6].
Chiral sulfenyl reagents
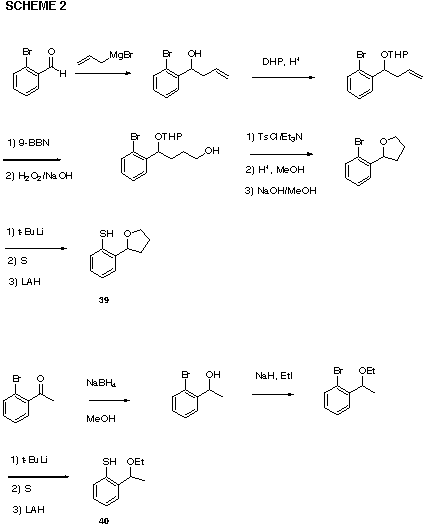
The pro-chiral reagents 39 and 40 were prepared from o-bromacetophenone and o-bromobenzaldehyde, correspondingly (Scheme 2). The addition reaction of 39 with 1-methylcyclohexene at ambient temperature produced only a 1.3:1 ratio of diastereomers as determined by integration of the proton nmr spectrum. Carrying out the reaction at &emdash;78�C served to increase the ratio to only 5:1. With styrene as the substrate the diastereomeric ratio of acetoxy sulfide products is only 1.44:1 with a chemical yield of 80%. With other substrates little or no selectivity was observed. Cyclization of substrate 31 initiated with the aryl sulfenyl chloride derived from 40 yielded a complex mixture with no clear evidence of diastereoselectivity.
It is not clear why the stereoselectivity associated with the sulfenyl halides is so much less than that observed with the corresponding selenenyl compounds [1]. The lack of selectivity may be a function of the smaller size of the sulfenyl cation compared to its selenenyl counterpart and its concomitant greater reactivity as an electrophile.
REFERNCES
1. W.H. Mueller, P. Batler, J. Am. Chem. Soc. 90,2075 (1968). See also [2] and [5]
2. Methoden Der Organischen Chemie, Band E11, 1985, Georg Thieme Verlag, Stuttgart-New York.
3. T. Wirth, Tetrahedron 55, 1 (1999) and references cited therein.
4. S. R. Harring, T. Livinghouse, Tetrahedron Lett. 30, 1499 (1989); C. Liu, K. Kudo, Y. Hashimoto, K. Saigo, J. Org. Chem. 61, 494 (1996); R. Deziel, E. Malenfant, C. Thibault, Tetrahedron Lett. 39, 5493
5. W.A. Smit, M.Z. Krimer, E.A. Vorob'eva, Tertahedron Lett., 2451 (1975)
6. H. Akita, T. Oishi, Chem.Pharm.Bull. 29, 1567 (1981); J. J. Parlow, Tetrahedron 50, 3297 (1994).
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0060 as the message subject of your e-mail.