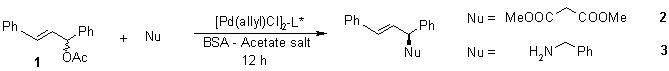
Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[A0062]
Enantioselective Palladium Catalyzed Allylic Substitution with New Multichiral Centers Monophosphine LigandsJean Michel Brunel, Martin Smith and Sebastien Reymond
Ecole Nationale Superieure de Syntheses, de Procedes et d’Ingenierie Chimiques d’Aix Marseille (E.n.s.S.P.I.c.a.m.), UMR 6516 du CNRS, Faculte de St Jerome, Av. Escadrille Normandie Niemen, 13397 Marseille, Cedex 20, FranceReceived: 2
August 2000 / Uploaded: 3 August 2000
In the last few years, there was a great interest in asymmetric variants of palladium catalyzed allylic reactions1. In this context, we have been witness to the design of chiral ligands capable of effective highly enantioselective reactions. Among them, C2 symmetric diphosphines2 and P,N-oxazolines3 have been throughly studied. However monophosphines or more generally ligands with one phosphorus linked to one or several heteroatoms, may also be useful. Thus, by contrast to chiral diphosphines, the field of chiral monophosphines has remained stagnant for a long time whereas numerous results in literature indicate that greater attention should be devoted to this family of compounds4. On the basis of such considerations, the synthesis of a new class of chiral monophosphine ligands and their ability to control enantioselective Pd-catalyzed allylic substitution has been investigated (Scheme 1).
Scheme 1
Contrarily to the methodology encountered in literature, we decided to prepare unusual multichiral center ligands bearing the chirality both at the phosphorus atom and at the chain. Synthesis of ligands 4-6 was readily achieved by an exchange reaction in refluxing toluene between tris(dimethylamino)phosphine and (S)-(+)-2-anilinomethylpyrrolidine followed by addition of the corresponding menthol derivative. The reaction was monitored by 31P NMR spectroscopy and after 1 hour the solvent was removed under vacuum. The products were purified by column chromatography affording the expected ligands 4-6 in good chemical yields varying from 52 to 70% and in a total anti diastereoselectivity (Scheme 2)5.

Scheme 2
We investigated the catalytic properties of the palladium complexes formed in situ from these ligands and [Pd(allyl)Cl]2 in an allylic alkylation of 1,3-diphenylprop-3-en-1-yl acetate 1 by the nucleophile generated from dimethylmalonate with N,O-bis(trimethylsilyl)acetamide (BSA) and a catalytic amount of an acetate salt (Table 1).
Table 1 : Enantioselective allylic alkylation of 1 with dimethylmalonate
Entrya |
Solvent |
Temperature (°C) |
Ratio 4/Pd |
Acetate salt |
Yield (%)b |
Ee (%)c |
1 |
THF |
25 |
4/1 |
AcOK |
14 |
41 (R) |
2 |
Toluene |
25 |
4/1 |
AcOK |
3 |
- |
3 |
CH2Cl2 |
25 |
4/1 |
AcOK |
90 |
89 (R) |
4 |
CH3CN |
25 |
4/1 |
AcOK |
80 |
80 (R) |
5 |
DMF |
25 |
4/1 |
AcOK |
65 |
45 (R) |
6 |
CH2Cl2 |
- 10 |
4/1 |
AcOK |
95 |
72 (R) |
7 |
CH2Cl2 |
25 |
4/1 |
AcONa |
95 |
79 (R) |
8 |
CH2Cl2 |
25 |
4/1 |
AcOAg |
94 |
66 (R) |
9 |
CH2Cl2 |
25 |
4/1 |
AcOK |
80 |
66 (R) |
10 |
CH2Cl2 |
25 |
1/1 |
AcOK |
90 |
51 (R) |
11 |
CH2Cl2 |
25 |
2/1 |
AcOK |
92 |
65 (R) |
12 |
CH2Cl2 |
25 |
3/1 |
AcOK |
91 |
76 (R) |
a
Experiments performed on a 0.39 mmole scale during 12 hours using 2 mol% of [{(h3-C3H5)PdCl}2]. b Isolated yield. c Ee measured on a Daicel Chiralcel OD-H column at l = 254 nm ; flow rate 1 mL/Min ; eluent : hexane/i-PrOH 200/1 , tR = 19.57 min, tS = 18.18 min.We first examined the use of ligand 4 in a variety of solvents, temperatures and ratios of ligand to palladium in allylic alkylation (Table 1). Dichloromethane appears to be the best solvent (entry 3). The lowering in enantioselectivity observed in changing to the more coordinating solvent DMF suggests that the ligand may be displaced by the solvent (entry 5, 45% ee). Lowering the temperature from 25 to -10°C does not improve the enantioselectivity (entries 3 and 6, 89% ee versus 72%, respectively). Increasing the ratio of 4/Pd from 1/1 to 1/4 improved the ee (51% ee versus 89% ee). Moreover, the influence of the acetate salt added has been studied and the best result was achieved using potassium acetate (entry 3, 89% ee) whereas sodium and silver acetate salts led to lower enantiomeric excesses (entries 7 and 8, 79 and 66% ee, respectively).
It is well known that the substrate leaving group or the counterion contained in the catalyst complex may influence the enantioselectivity of the reaction6. Thus, we decided to investigate the effect excerted by different anions on the enantioselectivity using various additive salts (Table 2).
Table 2 : Catalytic allylic alkylation of 4 in presence of various ammonium salts
Entrya |
Additive |
Ligand |
Yield (%)b |
Ee (%)c |
1 |
[NHex4]PF6 |
4 |
89 |
58 |
2 |
[NHex4]Cl |
4 |
88 |
74 |
3 |
[NHex4]BF4 |
4 |
86 |
62 |
4 |
[NHex4]Br |
4 |
92 |
84 |
5 |
[NHex4]I |
4 |
93 |
90 |
6 |
[NBu4]Br |
4 |
95 |
92 |
7 |
[NBu4]ClO4 |
4 |
85 |
85 |
8 |
- |
5 |
87 |
49 |
9 |
[NBu4]Br |
5 |
81 |
53 |
10 |
- |
6 |
85 |
41 |
11 |
[NBu4]Br |
6 |
91 |
56 |
a
Experiments performed on a 0.39 mmole scale during 12 hours using 2 mol% of [{(h3-C3H5)PdCl}2]. b Isolated yield. c Ee measured on a Daicel Chiralcel OD-H column at l = 254 nm ; flow rate 1 mL/Min ; eluent : hexane/i-PrOH 200/1 , tR = 19.57 min, tS = 18.18 min.Spectacular decreases or increases in the enantiomeric excesses have been observed depending on the nature of the counterion associated to the ammonium species. Thus, as depicted in Table 2, an important decrease of the enantioselectivity has been noted using PF6-, Cl- or BF4- anions whereas the presence of Br- or I- counterions improve slightly the enantiomeric excesses up to 92% ee. A similar improvement of the enantioselectivity using ligands 5 and 6 has been encountered when performing the reaction in the presence of tetrabutylammonium bromide salt (entries 9 and 11, 53 and 56% ee, respectively). On the other hand, enantioselectivity seems to be very sensitive to the structure of the ligand coordinated to the palladium. Thus, although minor structural modifications of 4 have been envisioned an important decrease of the enantiomeric excesses has been noticed in all cases (entries 8-11).
As an extension of this study, asymmetric allylic amination of 1,3-diphenylprop-3-en-1-yl acetate with benzylamine was carried out with palladium-ligand 4-6 complex catalysts (Table 3).
Table 3 : Enantioselective allylic amination of 1 with benzylamine
Entrya |
Solvent |
Temperature (°C) |
Ratio L*/Pd |
L* |
Yield (%)b |
Ee (%)c |
1 |
THF |
25 |
4/1 |
4 |
79 |
41 (S) |
2 |
Toluene |
25 |
4/1 |
4 |
5 |
- |
3 |
CH2Cl2 |
25 |
4/1 |
4 |
92 |
91 (S) |
4 |
CH2Cl2 |
- 10 |
4/1 |
4 |
95 |
78 (S) |
5 |
CH2Cl2 |
25 |
4/1 |
5 |
93 |
53 (S) |
6 |
CH2Cl2 |
25 |
4/1 |
6 |
94 |
56 (S) |
a
Experiments performed on a 0.39 mmole scale during 12 hours using 2 mol% of [{(h3-C3H5)PdCl}2]. b Isolated yield. c Ee measured on a Daicel Chiralcel OD-H column at l = 254 nm ; flow rate 1 mL/Min ; eluent : hexane/i-PrOH 200/1 , tR = 19.70 min, tS = 18.18 min.As already observed in the asymmetric allylic alkylation, the best experimental conditions have been encountered in CH2Cl2 at 25°C (entry 3, 91% ee). Moreover, ligands 5 and 6 provided level of enantioselectivities comparable ranging from 53-56% ee (entries 5 and 6) but lower that those previously obtained using ligand 4.
In conclusion, we have shown that readily accessible multichiral centers monophosphine compounds are efficient ligands for palladium catalyzed allylic substitutions. Further studies including modification to ligands design and mechanistic aspects are in progress.
Experimental section
Tetrahydrofuran (THF) and toluene were distilled from sodium/benzophenone ketyl immediately prior to use. Dichloromethane was distilled over P2O5. Ethylacetate and petroleum ether (35-60°C) were purchased from SDS and used without any previous purification. Column chromatography were performed on SDS silica gel (70-230 mesh). 1H and 13C NMR spectra were recorded in CDCl3 solution at 200.00 MHz and 50.30 MHz on a Bruker AC200 instrument, 31P NMR spectra were recorded in CDCl3 solution at 40.50 MHz on a Bruker AC100 (the usual abbreviations are used : s = singlet, d = doublet, t = triplet, q = quadruplet, m = multiplet). The positive chemical shift values are given in ppm, the coupling constants in Hertz. Specific rotations were determined with a Perkin Elmer polarimeter 341.
General procedure for the synthesis of ligands 4-5 :
Ligand 4 was prepared by exchange reaction at 110°C in 20 mL of anhydrous toluene between 163 mg of tris(dimethylamino)phosphine (1 mmole) and 176 mg of (S)-(+)-2-anilinomethylpyrrolidine (1 mmole) for 2 hours, followed by subsequent addition of 1 mmole of (-)-Menthol. Purification by flash chromatography using petroleum ether/ethylacetate (20/1) as eluent afforded ligand 4 as a white solid stable to air and moisture. Similar procedure was used in order to prepare ligands 5 and 6.
(2R,5S,9R,10R,13R)-2-menthoxy-3-phenyl-1,3-diazaphosphabicyclo[3.3.0]octane 4
70% yield ; M.p. 83°C; [a]D20 = - 330.4 (c = 1.02, CH2Cl2); 31P NMR (40.5 MHz, CDCl3): d = 128.5; 1H NMR (200 MHz, CDCl3): d = 7.25-7.15 (m, 2H), 7.02-6.98 (m, 2H), 6.80 (t, J = 7.2 Hz,1H), 4.12 (qt, J = 6.2 Hz, 1H), 3.81-3.40 (m, 3H), 3.25-3.08 (m, 2H), 2.14-1.56 (m, 8H), 1.55-0.70 (m, 11H); 0.60 (d, J = 6.9 Hz, 3H); 13C NMR (50 MHz, CDCl3): d = 145.9 (d, J = 15.9 Hz), 129.0 (2C), 118.7, 115.2 (d, J = 13.3 Hz, 2C), 74.5 (d, J = 9.6 Hz), 62.9 (d, J = 7.9 Hz), 53.6 (d, J = 7.1 Hz), 49.1 (d, J = 2.5 Hz), 48.5 (d, J = 36.3 Hz), 44.0, 34.4, 32.1, 32.0, 26.3 (d, J = 4.3 Hz), 25.1, 23.0, 22.3, 21.3, 15.4; C21H33N2OP (360.23): calcd. C 70.0, H 9.2, N 7.8, P 8.6; found C 69.8, H 9.5, N 7.6, P 8.8.
(2R,5S,9S,10R,13S)-2-menthoxy-3-phenyl-1,3-diazaphosphabicyclo[3.3.0]octane 5
58% yield ; M.p. 96°C; [a]D20 = - 255.5 (c = 1.01, CH2Cl2); 31P NMR (40.5 MHz, CDCl3): d = 123.7; 1H NMR (200 MHz, CDCl3): d = 7.29-7.21 (m, 2H), 7.07-7.03 (m, 2H), 6.88-6.84 (t, J = 7.2 Hz, 1H), 4.25-4.10 (qt, J = 6 Hz, 1H), 3.85-3.45 (m, 3H), 3.27-3.15 (m, 2H), 2.30-1.00 (m, 13H), 0.85 (d, J = 5.7 Hz, 3H), 0.79 (d, J = 6.6 Hz, 3H), 0.73 (d, J = 6.9 Hz, 3H); 13C NMR (50 MHz, CDCl3): d = 146.0, 129.0 (2C), 118.7, 115.2 (d, J = 12.1 Hz, 2C), 74.3, 63.5 (d, J = 8.7 Hz), 53.9 (d, J = 7.3 Hz), 49.1 (d, J = 4.2 Hz) , 48.4 (d, J = 37.9 Hz),43.7, 34.5, 32.3, 32.0, 26.3 (d, J = 4.2 Hz), 25.3, 22.3, 21.3, 15.6; C21H33N2OP (360.23): calcd. C 70.0, H 9.2, N 7.8, P 8.6; found C 70.1, H 9.2, N 8.0, P 8.4.
(2R,5S,9R,10R,13R)-10-(phenylmenthoxy)-3-phenyl-1,3-diazaphosphabicyclo[3.3.0]octane 6
52% yield ; 31P NMR (40.5 MHz, CDCl3): d = 130.5; 1H NMR (200 MHz, CDCl3): d = 7.26-6.97 (m, 9H), 6.83-6.67 (t, J = 7.2 Hz, 1H), 4.05—3.92 (m, 1H), 3.81-3.45 (m, 2H), 3.27-3.17 (m, 2H), 2.0-0.8 (m, 13H), 1.45 (s, 3H), 1.32 (s, 3H); 0.73 (d, J = 6.2 Hz, 3H); 13C NMR (50 MHz, CDCl3): d = 151.6, 145.9 (d, J = 13.4 Hz), 129.0 (2C), 127.8 (2C), 125.9 (2C), 125.4 (d, J = 33.5 Hz), 118.9, 115.5 (d, J = 13.0 Hz), 75.4 (d, J = 14.8 Hz), 63.6 (d, J = 7.8 Hz), 52.7 (d, J = 7.1 Hz), 52.3 (d, J = 4.5 Hz), 48.7 (d, J = 36.1 Hz), 44.8 (d, J = 2.9 Hz), 40.9, 34.7, 32.3, 31.8, 30.7, 27.7, 26.3 (d, J = 4.3 Hz), 22.4, 21.9; C27H37N2OP (422.25): calcd. C 73.9, H 8.4, N 6.6, P 7.3; found C 74.0, H 8.2, N 6.9, P 7.1.
General procedure for palladium catalyzed allylic alkylation (Table 1, entry 3):
A mixture of [Pd(allyl)Cl]2 (3 mg, 8.2 10-3 mmol (2 mol%)) and the ligand 4 (12 mg, 33 10-3 mmol) in anhydrous CH2Cl2 was stirred at room temperature for 15 minutes. A solution of 1,3-diphenylprop-3-en-1-yl acetate (100 mg, 0.396 mmol) was added and stirring was maintained for 5 minutes. Dimethylmalonate (157 mg, 1.18 mmol), N,O-bis(trimethylsilyl)acetamide (BSA, 241 mg, 1.18 mmol) and a catalytic amount of potassium acetate (KOAc) were subsequently added. The resulting solution was stirred at 25°C for 12 hours . The solution was diluted with Et2O (3x10mL). The combined organic phases were dried over MgSO4 and filtered. The solvent was removed in vacuo to afford a pale yellow oil that solidified on standing.The enantiomeric excesses can be determined on the crude mixture by HPLC analysis on a Daicel Chiralcel OD-H column (l = 254 nm ; flow rate 1 mL/Min ; eluent : hexane/i-PrOH 200/1 , tR = 19.57 min, tS = 18.18 min).
General procedure for palladium catalyzed allylic amination (Table 3, entry 3):
A mixture of [Pd(allyl)Cl]2 (3 mg, 8.2 10-3 mmol (2 mol%)) and the ligand 4 (12 mg, 33 10-3 mmol) in anhydrous CH2Cl2 was stirred at room temperature for 15 minutes. A solution of 1,3-diphenylprop-3-en-1-yl acetate (100 mg, 0.396 mmol) was added and stirring was maintained for 5 minutes. Benzylamine (126 mg, 1.18 mmol) was subsequently added. The resulting solution was stirred at 25°C for 12 hours . The solution was diluted with Et2O (3x10mL). The combined organic phases were dried over MgSO4 and filtered. The solvent was removed in vacuo to afford a pale yellow oil.The enantiomeric excesses can be determined on the crude mixture by HPLC analysis on a Daicel Chiralcel OD-H column (l = 254 nm ; flow rate 1 mL/Min ; eluent : hexane/i-PrOH 200/1 , tR = 19.7 min, tS = 21.70 min).
Notes and References.
(2) For example : (a) Trost, B. M. ; Organ, M. G. ; O’doherty, G. A. J. Am. Chem. Soc. 1995, 117, 9662-9670. (b) Bolm, C. ; Kaufmann, D. ; Gessler, S. ; Harms, K. J. Organomet. Chem. 1995, 502, 47-52. (c) Seebach, D. ; Devaquet, E. ; Ernst, A. ; Hayakawa, M. ; Kuhnle, F. N. M. ; Schweizer, W. B. ; Weber, B. Helv. Chim. Acta 1995, 78, 1636-1650.
(3) (a) von Matt, P. ; Pfaltz, A. Angew. Chem., Int. Ed. Engl. 1993, 32, 566-569. (b) Sprinz, J. ; Helmchen, G. Tetrahedron Lett. 1993, 34, 1769-1773. (c) Dawson, G. J. ; Frost, C. G. ; Williams, J. M. J. ; Coote, S. J. Tetrahedron Lett. 1993, 34, 3149-3150.
(4) Lagasse, F. ; Kagan, H. B. Chem. Pharm. Bull. 2000, 48, 315-324.
(5) The notations of the syn and anti diastereomers are according to the methylene substituent of the pyrrolidine ring with respect to the extracyclic aryl group. If they are at the same side of the five membered phosphorus-containing ring, we call it a syn diastereomer; otherwise, it is an anti diastereomer. (a) Cros, P.; Buono, G. ; Peiffer, G. ; Denis, D. ; Mortreux, A. ; Petit, F. New. J. Chem. 1987, 11, 573. (b) Brunel, J. M. ; Chiodi, O. ; Faure, B. ; Fotiadu, F. ; Buono, G. J. Organomet. Chem. 1997, 529, 285.
(6) For a detailed investigation and interpretation of this particular aspect, see : Burckhardt, U. ; Baumann, M. ; Togni, A. Tetrahedron Asymmetry 1997, 8, 155-159 and references cited therein.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0062 as the message subject of your e-mail.