
[A0072]
The Effect of p-Bond Screening on the Reaction of s-Tetrazines
with
7-Substituted Benzonorbornadienes
Ronald N. Warrener* and Peter A. Harrison
Centre for Molecular Architecture,
Central Queensland University, Rockhampton, Queensland, 4702, Australia,
E-mail: [email protected]
Received: 4 August 2000 / Uploaded: 5 August 2000
The use of norbornadiene 1 as a transfer reagent for acetylene is well established1 and its reaction with s-tetrazines is representative. This is now an established method for the conversion of s-tetrazines to pyridazines where norbornadiene provides the C4 and C5 atoms of the pyridazine ring.2 The reaction of norbornadiene 1 with 3,6-di(2-pyridyl)s-tetrazine 2 is illustrated in Scheme 1 and involves the following Diels-Alder sequence: Diels-Alder addition of 2 onto 1 to form the transient, primary intermediate 3 which instantaneously collapses by retro-Diels-Alder elimination of dinitrogen, to afford the dihydropyridazine 4. A second retro-Diels-Alder reaction of 4 yields cyclopentadiene 5 and the pyridazine 6. The reaction is extremely facile with the diene 4 being formed rapidly at room temperature and complete fragmentation to 5 and 6 can be achieved by gentle heating (50-60 oC) for a few minutes. While the pyridazine 6 is the target in this reaction, it is very often the other fragmentation partner which is the more appealing. Thus, replacement of the 7-methano carbon of norbornadiene with hetero atoms 9 has been used to form substituted furans 10 or pyrroles 11, and replacement with a vinylidene bridge allows access to fulvenes, eg 12.3 This is especially useful for the preparation of 3,4-disustituted derivatives of these ring-systems where the short sequence from the unsubstituted ring 7 to the substituted compounds 10-12 is shown Scheme 1.
Scheme 1
The same cycloaddition/fragmentation sequence can be applied to the benzoderivatives 13 by treatment with s-tetrazine 2 to offer entry to isobenzofurans 15a,4 isoindoles 15b5 and isobenzofulvenes 15c6 (Scheme 2) under the same mild reaction conditions. The notable exception is benzonorbornadiene 21, which still reacts with the s-tetrazine 2 to form the dihydropyridazine intermediate 28, but fails to undergo the fragmentation step to form isoindenes. However, it is this resistance to fragmentation and the potential of the conjugated dihydropyridazines as Diels-Alder 1,3-dienes which made it attractive to investigate this reaction in more detail .
Scheme 2

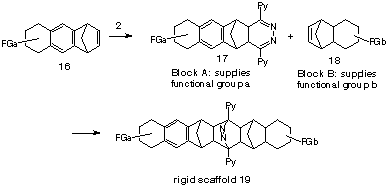
This interest in such 1,3-dienes is associated with the ease of reaction and the range of alkenes (notional dienophiles) which can be converted in one step to dihydropyridazines (notional 1,3-dienes). Indeed, by reaction of a norbornene substrate with half an equivalent of s-tetrazine, it is possible to form the dihydropyridazine1,3-diene in the presence of the unreacted dienophile, thereby setting up the opportunity to form a symmetrical adduct by Diels-Alder addition of the two partners. In this way, the s-tetrazines 2 has acted as a molecular glue to join functionalised norbornene substrates (building blocks),7 in another version of our molecular 'lego',8 as a route to bridged scaffolds (Scheme 3). In this process, a prefabricated building block, eg 16 with a terminal norbornene p-bond is converted to its fused dihydropyridazine, eg 17 prior to coupling with a second building block, eg 18 to form a mixed coupled product, eg 19. The practical requirement is the first-formed 4,5-dihydropyridazine must have sufficient lifetime to react with the second norbornene-type functionalised block in the coupling step to form the product 13. Accordingly, norbornadienes 1 or 7-oxabenzonorbornadienes 13, which display lability towards fragmentation, can only be used in the second step of the coupling process. Our interest in using benzonorbornadienes in either step revolved about their potential to act as delivery agent for various types of functionality which could be attached to the benzene ring. In this respect, we required detail of their reaction with s-tetrazines so that they could be assessed as reagents.
Scheme 3
In this report, we describe the reaction of s-tetrazine 2 with benzonorbornadiene 21 and some of its derivatives 23, 25 and 27, where the 7-methano bridge of 21 have been modified by alkyl or ring substituents. The other feature of this study was to establish the effect that modification of the 7-substituent would have on the facial selectivity of attack at the norbornene p-bond and to assess this as a geometry-controlling element in the 'lego' block building program.
At the commencement of this program, benzonorbornadiene 21 and the spirocyclopropyl derivative 23 had been reported (Scheme 4). The 7,7-dimethyl-derivative 25 was prepared using similar methodology by reacting 5,5-dimethylcyclopentadiene9with benzyne and isolated as a liquid. The 1H n.m.r. spectrum was very simple reflecting the expected Cs-symmetry of 25: singlets at d 0.79, 1.25 for the methyl protons, with the higher-field resonance being ascribed to the methyl group under the shielding influence of the aromatic ring; coupled resonances between the bridgehead protons (d 3.34) and for the vinyl protons (d 6.65) and two sets of finger-like quartets for the aromatic protons at d 6.8-7.2 are entirely in congruence with the assigned structure.
Scheme 4

The spirocyclopentyl derivative 27 was prepared from spiro[4,4]nona-1,3-diene 2610 and benzyne in a similar fashion and it also was a liquid. The 1H n.m.r. spectrum was essentialy identical with 25 except that the singlet methyl protons were replaced with a broad multiplet at d 1.1-1.9 for the cyclopentyl methylene protons.
Scheme 5
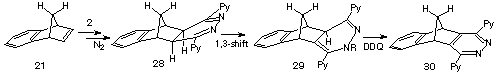
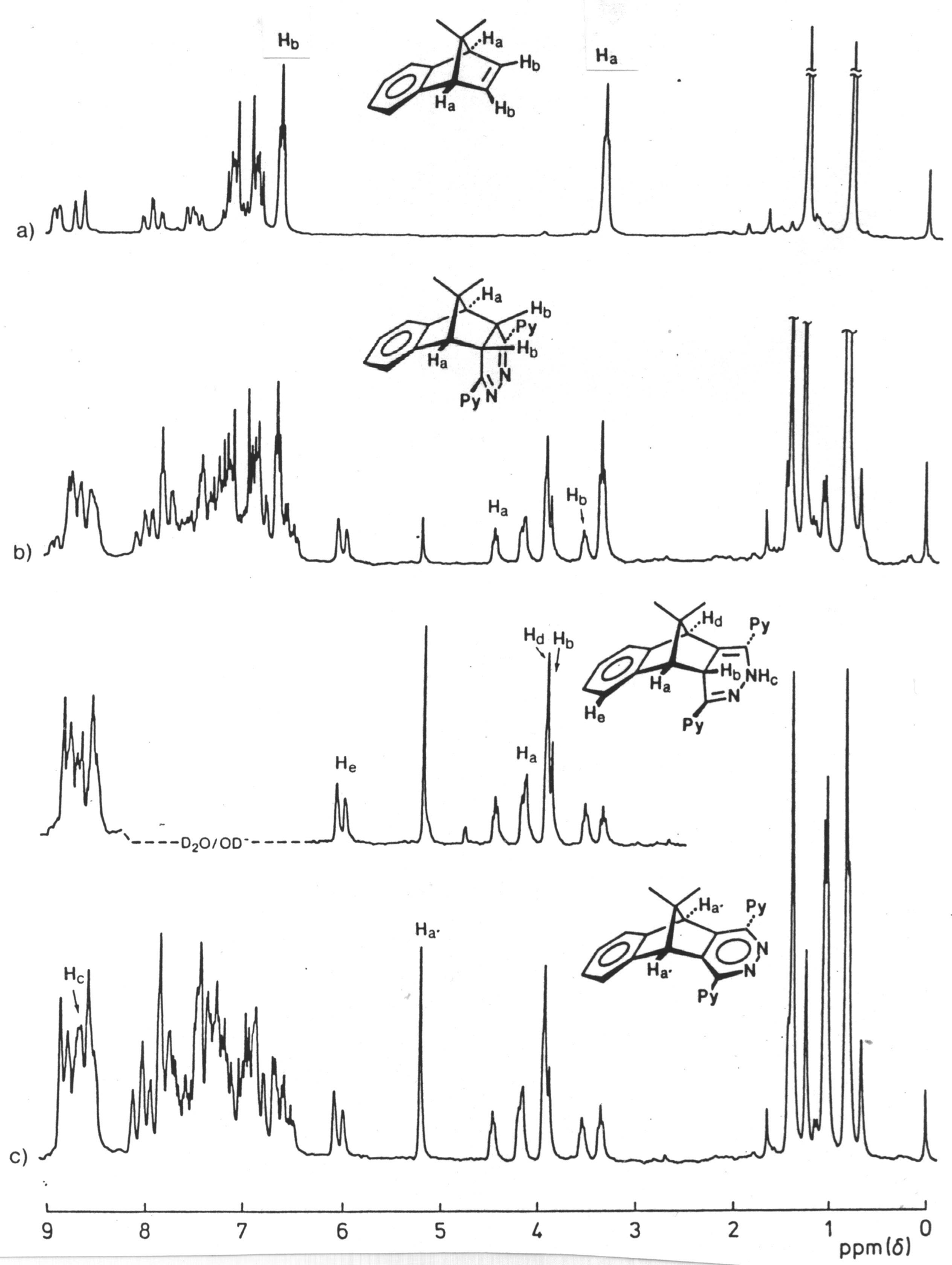
The reaction of the s-tetrazine 2 with benzonorbornadiene 21 (Scheme 5) was conducted in chloroform at 30 oC and monitored by 1H n.m.r. spectroscopy ; three traces are shown in Figure 1. This indicated that significant reaction has occurred after 10 min (Figure 1, trace 1a) to produce a single compound characterised by having singlet resonances at d 3.58 and 3.79 assigned to the methine protons Ha, Hb of the 4,5-dihydropyridazine 28. While conversion of 21 to 28 was complete after 30 min, evidence for the formation of a second product had emerged (Figure 1, trace 1b) and become the almost exclusive product after 2 hours (Figure 1, trace c). This new product has lost the Cs symmetry of its precursor 28 and was assigned the rearranged 1,4-dihydropyridazine structure 29; there is good precedent for this rearrangement.11 It was characterised by the presence of four singlets d 9.27, 4.36, 4.67, 2.67 where the low-field resonance is assigned to the NH proton, the presence of which was already confirmed by i.r. spectroscopy (nmax 3350 cm-1), and this assignment is supported by exchange with DO- in D2O. The resonance at d 2.76 is attributed to the Hb proton of the heterocyclic ring which might be expected to resonate at much lower field as it is doubly allylic, so clearly an upfield component is also operative.
Figure 1. 1H nmr
spectra (80 MHz; CDCl3) of products resulting from the reaction between
s-tetrazine 2 and benzonorbornadiene 21 after
a)10 min at 30 oC b) 30 min at 30 oC c) 120 min at 30 oC
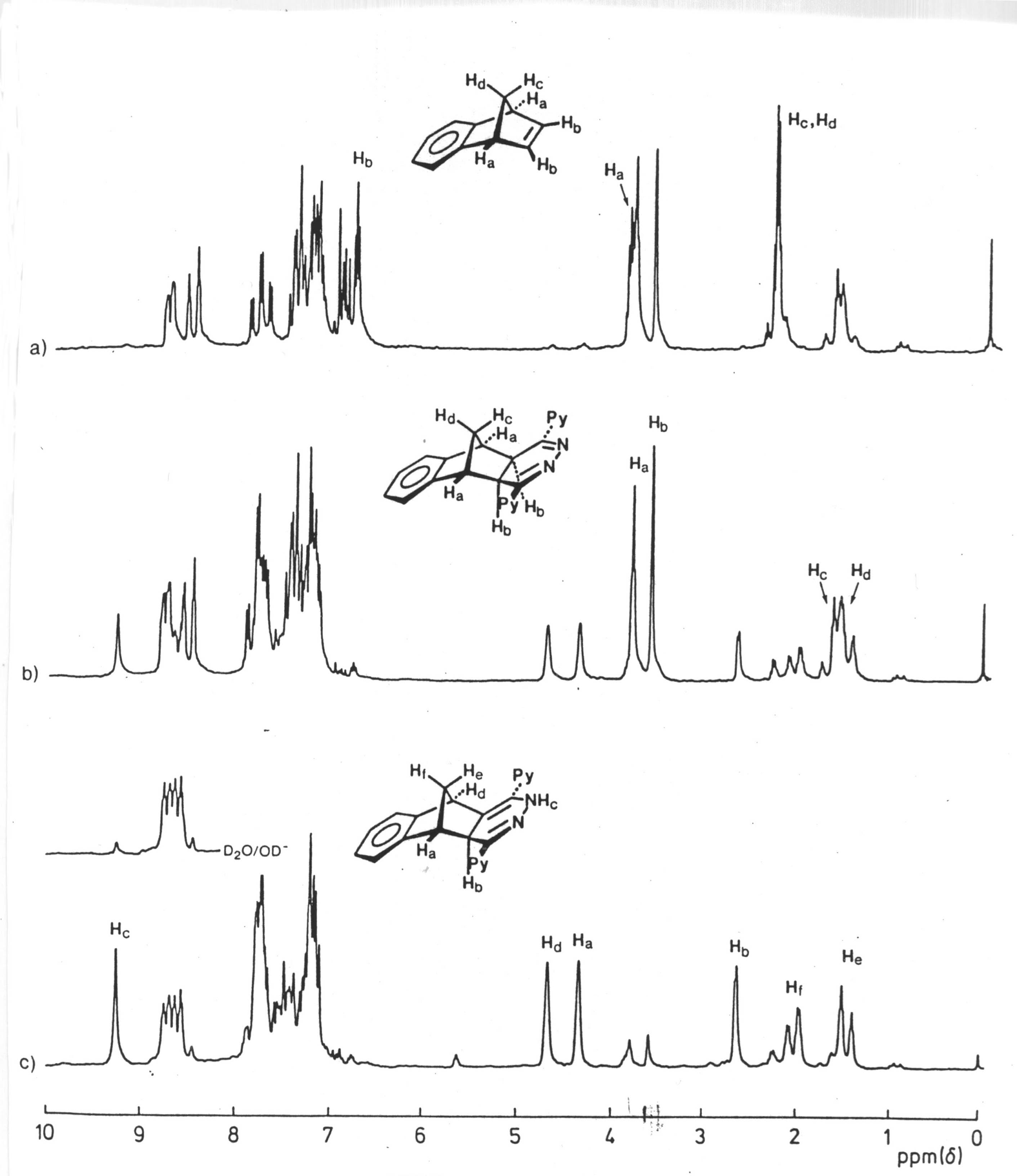
Figure 2. Stereoview of preferred conformation of 29 determined by AM1.

Molecular modelling (AM1, Figure 2) of 29 indicated that the pyridyl ring vicinal to Hb and the phenylene ring each offer shielding to Hb which accounted for its upfield shift. This result also offered indirect confirmation of the exo fusion in 29 and this was supported by the lack of coupling between Hb and the vicinal bridgehead proton Ha.
The commonality of basic ring-structures in 28 and 29 was confirmed by oxidation of mixtures of these products with dichlorodicyanoquinone (DDQ) to the same Cs-symmetric pyridazine 30.
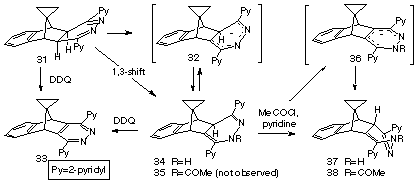
When the spirocyclopropylbenzonorbornadiene 23 was treated with s-tetrazine 2 (Scheme 6), only slow evolution of nitrogen was observed. The 4,5-dihydropyridazine intermediate 31 was none-the-less the first-formed product as deduced from the time-dependent 1H n.m.r. study shown in Figure 3. The structure of the fused 4,5-dihydropyridazine 31 was again indicated by the presence of two singlet methine resonances at d 3.21 and 3.67 which confirmed both the Cs symmetry of the product and its exostereochemistry (no 3J-coupling). Furthermore, under normal reaction conditions (56 oC), this intermediate did not fragment further to yield the respective isoindene, but preferentially underwent rearrangement.
Figure 3 1H nmr spectra (80 MHz;
CDCl3) of products resulting from the reaction between
s-tetrazine 2 and 7-spirocyclopropylbenzonorbornadiene 23
after
a) 30 min at 30 oC b) 120 min at 30 oC c) 24 hours at 60 oC

The fact that rearrangement product 34 retained the benzonorbornanyl ring-structure present in its precursor 31, was established by oxidation with DDQ,12 where the same pyridazine 33, mass spectrum (M+ m/z 374), was produced from enriched mixtures favouring each product. The 1H n.m.r. of 33 was consistent with that expected for a structure of high symmetry (Figure 4).
The structure of 1,4-dihydropyridazine 34 was assigned very much by analogy with 29 and in an attempt to form a derivative suitable for characterisation, it was treated with acetyl chloride in pyridine at reflux overnight.
Figure 4. 1H nmr spectrum (80 MHz; CDCl3) of pyridazine 33

The isolated product had the expected mass spectral peak at m/z = 418 for an acetyl derivative and an M-43 peak, corresponding to a loss of -COCH3, the 1H n.m.r. spectrum (Figure 5) indicated that rearrangement had occurred. The fact that there was vicinal coupling between protons Ha and Hb (d 4.0, d 4.1 J = 3.8 Hz) excluded the structure 35 (endo-Hb) and supported the structure 38 (exo-Hb). This assignment was also supported by the increased dispersion of the aromatic resonances, a manifestation of the proximity of the phenyl and one of the pyridyl rings.13 The expected acetate methyl group occurred as a characteristic, sharp singlet (d 2.32).
A clue to the mechanism for inversion of stereochemistry which occurred during the acetylation, is apparent in the deuterium exchange spectrum (Figure 3, trace c and above) of the starting material 35 where Hb was exchanged under the basic conditions as well as the NH proton. This implicated the anion 36, demonstrating the acidity of the methine proton at the bridgehead of the heterocyclic ring. Accordingly, formation of the acetate 35 may well occur but isomerisation to the observed product would ensue since formation of anion 36 would be favoured under the basic conditions of the reaction which is conducted in pyridine at reflux, and protonation from the favoured exo-face would yield the observed product 38.
Figure 5. 1H nmr spectrum (80 MHz; CDCl3) of N-acetyl 1,4-dihydropyridazine 38

Scheme 6
The reaction between benzonorbornadiene 21 and s-tetrazine 2, which was essentially complete after 2 hours at room temperature, occurred at least an order of magnitude faster than that observed for spirocyclopropyl derivative 23. What, then, if the norbornene p-bond was even more screened by the 7-substituents?
Figure 6. 1H n.m.r spectra (80 MHz;
CDCl3) of the reaction between
s-tetrazine 2 and 7,7-dimethyl benzonorbornadiene 25 after
a) 1 hour at 30 oC; b) 12 hours at 60 oC; c) 36 hours at 60oC
The time-dependent 1H n.m.r. study of the reaction of s-tetrazine 2 with the 7,7-dimethylbenzonorbornadiene 25 (Figure 6) confirmed that reaction was even slower, with essentially no change having occurred at room temperature after 12 hours (Figure 5, trace a). Increasing the temperature to 56 oC confirmed that reaction would proceed, but there were differences. First, the reaction yielded the dehydrogenated pyridazine 41 as the major reaction product after 36 hours (Figure 5, trace c). Secondly, the spectral data indicated that initial attack had occurred from the endo-face of 25.
Figure 7 1H nmr spectra (80 MHz;
CDCl3) of products resulting from the reaction between
s-tetrazine 2 and 7-spirocyclopentylbenzonorbornadiene 27
after
a) 1 hour at 30 oC b) 12 hours at 60 oC c) 36 hours at 60 oC

There was also evidence to support an apparent equilibrium between symmetric dihydropyridazine 39 and the unsymmetrical isomer 40 being maintained prior to dehydrogenation to the pyridazine 41. The resonance ascribed to exo-protons Hb (annotated in Figure 5) in either dihydropyridazine 39 or 40 showed no evidence for deuterium exchange which reflected their crowded environment as expected if the exo-stereochemical assignment was correctly assigned.
This change from exo-attack on 23 to endo-attack on 25 by s-tetrazine 2 was entirely in keeping with the known ability of syn-substituents on the methano-bridge to screen the p-bond, and has been observed in other systems. Thus, it was not surprising to find that the predicted endo-attack for the reaction of 2 with the spirocyclopentylbenzonorbornadiene 27 was also observed in practice. The slow rate of attack at the p-bond of 27 by 2 was very similar to that for the 7,7-dimethyl derivative 25, but conversion of the intermediate dihydropyridazines 42 and 43 to the pyridazine 44 was even faster. The time-dependent 1H n.m.r. study (Figure 7) provided evidence for endo-attack of 2 at the p-bond of 27, and was again based on the coupling between Ha and Hb (see annotations for relevant proton assignments). Rearrangement to the 1,4-isomer was accompanied by substantial oxidation to the pyridazine 44 upon raising the temperature to 60 oC, however, oxidation was still incomplete after 36 hours.
Scheme 7.

Conclusion:
The attack of s-tetrazine 2 on the p-bond of benzonorbornedienes is markedly influenced by the substituents at the 7-position. Exo-attack occurred with the parent system 21, and a similar facial selectivity was observed with the spirocyclobenzonorbornadiene 23, however the reaction was much slower. Introduction of the spirocyclopentyl at the 7-position screened the exo-face from attack and forced endo-facial attack to occur; a similar result was obtained with the gem-dimethyl derivative 25. The initially-formed 1,3-dihydropyridazines were unstable and were, in all cases, transformed by prototropic isomerisation to their 1,4-isomers, which dehydrogenated to the related pyridazine under the reaction conditions in compounds with the endo-precursors, or by specific dehydrogenation (DDQ) in the case of the exo-products.
Experimental
All melting points were determined on a Reichert hot-stage microscope, and are uncorrected. Microanalyses were performed by the Australian National University Microanalytical Service. Ultraviolet spectra were recorded on a Unicam SP800 spectrophotometer, using matched 5 mm or 10 mm silica cells. Infrared spectra were recorded on either a Unicam SP200G spectrophotometer or a Perkin-Elmer 283 spectrophotometer. Unless otherwise specified, infrared spectra were obtained using a Nujol mull between sodium chloride discs. Where spectra were recorded on solutions, they were obtained using 1 mm sodium chloride cavity cells. The intensities of i.r. absorptions are reported as s (strong, > 50% intensity of maximum absorption), m (medium, 25-50% max.) or w (weak, 12-25% max.). 1H n.m.r. spectra were recorded on a Varian CFT20 (80 MHz, Fourier mode), a Jeol JNM-MH-100 (100 MHz, continuous wave), or a Brucker HFX-270 (270 MHz, Fourier mode). 13C n.m.r. spectra were recorded on a Jeol JNM-FX-60 (15.04 MHz), a Varian CFT-20 (20.00 MHz) or a Brucker HFX-270 (67.89 MHz) n.m.r. spectrometer. 1H and 13C n.m.r. spectra were obtained using solutions in 5 mm and 10 mm tubes respectively, with tetramethylsilane (TMS) as internal standard. All chemical shifts are expressed in parts per million (ppm) downfield from TMS (d scale). Coupling constants (J) are given in Hertz, with multiplicity patterns designated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) and br (broad). Simulated spectra were calculated using the NTCSIM or ITRCAL procedures, available as part of the Nicolet 1180 Fourier Package. Low resolution mass spectra were recorded on either a Varian MAT CH7 or an A.E.I. MS902 mass spectrometer. The latter instrument was used for high resolution mass measurements. Unless otherwise stated, all peaks greater than 5% of the intensity of the base peak are reported. Combined GC-MS was carried out using a Varian MAT 111 (0.125 inch column, 2% OV-17) system. Gas chromatographic analyses, and preparative separations were performed on either a Hewlet-Packard Model 5754B (12 ft x 0.25 inch metal column, 10% SE 30 on 60-100 mesh Embacel) or a Packard 7400 Series (2 m x 1 cm glass column, 10% SE 30 on 44-60 mesh Embacel) instrument. A Waters Associates Series 6000 system was used for analytical HPLC. Preparative thinlayer chromatography was carried out on 20 x 100 cm glass plates using silica gel (Merck HP254 as absorbent, or on 20 x 20 cm Merck precoated (2 mm, 60 F254) PLC plates. Either Spence Type H activated alumina or May and Baker chromatography silica gel was employed in column chromatography.
l',4'-Dihydrospiro[cyclopentane-l,9'-[l,4]methononaphthalene] (27)
Spiro[4,4]nona-l, 3-diene 2610 (40 g, 0.33 mol) was added to a solution of isoamylnitrite (58 g, 66 ml, 0.5 mol) in methylene chloride (400 ml). The resulting solution was heated to reflux and treated slowly, over 2 h, with a solution of anthranilic acid (68.6 g, 0.5 mol) in the minimum volume of acetone (ca. 300 ml). The mixture was maintained under reflux for a further 2 h, and the bulk of the solvent removed by distillation. Water (500 ml) was added, and the resulting mixture extracted with light petroleum (2 x 400 ml). The combined organic extracts were washed with 5% aq. NaOH (3 x 500 ml) and brine (2 x 500 ml), and dried. The solvent was removed under reduced pressure and the residual brown coloured liquid purified by distillation. The product 27 was collected at 80-87 oC/ 0.5 mbar as a colourless liquid. The distillation residue was chromatographed on a column of alumina (6 cm x 20 cm), using light petroleum as eluent, to yield a further portion of the product 27 (total yield, 32.3 g, 50%). An analytical sample of the title compound 27 was obtained by short path distillation (50 oC / 0.1 mbar).
(Found: C, 91.9; H, 8.2. C15H16 requires C, 91.8; H, 8.2%). U.v. lmax (e) 264 (560), 270 (600), 272 infl. (540), 278 (480) nm. I.r. nmax(liquid film) 3070s, 3050m, 3020m, 2970s, 2870s, 1455s, 1325w, 1300m, 1245w, 1195w, 1140w, 1010w, 925w, 900w, 855w, 790s, 740s, 695s, 665m cm.-1 1H n.m.r. spectrum, (80 MHz, CDCl3): d 1.1-1.9 (8H, m, cyclopentane protons), 3.46 (2H, dd Ja, b=2.0 Hz, Ja, b'=2.0 Hz, Ha), 6.70 (2H, dd Ja, b =2.0 Hz, Ja', b =2.0 Hz, Hb), 6.8-7.2 (4H, m, aromatic protons). 13C-{1H} n.m.r. spectrum, (20 MHz, CDCl3): d 25.3 (C3, C4), 33.3 / 33.8 (C2,C5), 59.1 (C1', C4'), 91.1 (C1), 121.8 / 124.0 (C5', C6', C7', C8'), 142.5 (C2', C3'), 151.9 (C4'a, C8'a). Mass spectrum: m/z (%) 197 (18), 196 (M+, 100), 195 (10), 181 (50), 167 (72), 166 (17), 165 (28), 155 (34), 154 (50), 153 (37), 152 (25), 142 (27), 141 (42), 129 (14), 128 (59), 127 (11), 115 (20), 39 (12).
1,4-dihydro-9,9-dimethyl-1,4-methanonaphthalene (25).
Compound 25 was prepared in the same way as 27, in this case from 5,5-dimethylcyclopentadiene 249 by reaction with benzyne 20. The title compound 25 was separated by preparative tlc (silica / n-hexane) and purified by short-path distillation (50 oC / l0-2 mbar), as a colourless liquid (40%). (Found, C, 91.8; H, 8.3. Cl3Hl4 requires C, 91.7; H, 8.3%). U.v. lmax (e) 263 (580), 271 (610), 273 infl. (540), 279 (480) nm. I.r. nmax (liquid film) 3070s, 3020m, 2970s, 2930s, 2910m, 2870s, 1470m, 1455s, 1445m, 1385m, 1365m, 1300m, 1265m, 1220w, 1135m, l0l0w, 925w, 915w, 855w, 790s, 740s, 700s, 670m cm-1 1H n.m.r. spectrum (80 MHz, CDCl3): d 0.79 (3H, s, anti-CH3), 1.25 (3H, s, syn CH3), 3.34 (2H, dd Ja, b=2.1 Hz, Ja,b'=2.1 Hz, Ha), 6.65 (2H, dd Ja, b=2.1 Hz, Ja', b=2.1 Hz, Hb), 6.8-7.2 (4H, AA'BB' pattern, aromatic protons). 13C-{1H } n.m.r. spectrum (20 MHz, CDCl3): d 22.8 / 23.4 (C10, C11), 60.2 (C1, C4), 78.6 (C9), 122.1 (C5, C8), 124.0 (C6, C7), 141.8 (C2, C3), 151.6 (C4a, C8a). Mass spectrum: m/z 170 (M+, 30%), 156 (14), 155 (100), 153 (14), 128 (18).
Reaction of 1',4'-dihydro-spiro[cyclopropane-1,9'-[1,4]methanonaphthalene] (23) with 3,6-di(2'-pyridyl)-s-tetrazine (2).
A solution of the olefin (23) (2.5 g, 14.9 mmol) and s-tetrazine (2) (3.6 g, 14.9 mmol) in chloroform (10 ml) was heated under reflux overnight. The resulting solution was filtered through a bed of alumina and freed of solvent to yield the crude, crystalline product mixture. Recrystallization from benzene / n-hexane afforded a mixture of (cis-4'a-cisoid4'a, 5'-cis-5')-1', 2": 4', 2"-dipyridyl4'a, 5', 10', 10'a-tetrahydrospiro[cyclopropane-1,11'-[5, 10]methano benzo[g]phthalazine (31) and (cisoid-4'a,5'-cis-5')-1',2":4',2"'dipyridyl-4'a, 5', 10'-tetrahydro spiro[cyclopropane-1,11'[5,10]methanobenzo[g]phthalazine] (34) (ratio ca 1:4) (4.6 g, 83%) as buff coloured prisms. (Found: M+, 376.1687. C25H20N4 requires 376.1688) I.r. vmax 3350m (>NH) cm.-1. 1H n.m.r. spectrum (80 MHz, CDCl3): d -0.2-0.6 (m, cyclopropane protons), 2.76 (s, Hb), 3.21 (s, Ha), 3.64 (s, Ha'), 3.67 (s, Hb), 4.14 (s, Hd'), 7.0-8.7 (m, aromatic protons), 9.14 (br s, exchanged with DO- / D2O, Hc'). Mass spectrum: m/z 376 (M+, 70%) , 299 (58), 272 (72), 142 (31), 78 (100).
Preparation of (cis-4'a-transoid-4'a,5'-cis-5')-1',2":4',2"'-dipyridyl-4'a,5',10',10'a-tetrahydrospiro[cyclopentane-1,11'-[5,10]methanobenzo[g]phthalazine] (42) and (transoid-4'a,5'-cis-5')-1',2":4',2"-dipyridyl-2',4'a,5',10'-tetrahydrospiro[cyclopropane-1,11'-[5,10]methanobenzo[g]phthalazine] (43).
Similar treatment of 1', 4'-dihydrospiro[cyclopentane-l, 9'-[1,4]methano naphthalene] (27) with s-tetrazine (2) afforded a mixture of (42) and (43) The mixture was not purified, and characterised only by 1H n.m.r. spectroscopy. 1H n.m.r. spectrum, (80 MHz, CDCl3): d 1.0-2.0 (m, cyclopentane protons), 3.61 (dd Ja b= 2.1 Hz, Ja', b= 2.1 Hz, Hb), 3.80 (d Ja' b'=3-5 Hz, Hb'), 4.03 (s, Hd'), 4.25 (dd Ja' b'= 3.5 Hz, Ja' d'= 1 4 Hz, Ha'), 4.33 (dd Ja b= 2.1 Hz, Ja b'= 2.1 Hz, Ha), 6.0-9.0 (m, aromatic protons), 8.59 (s, exchanged with DO-/ D2O, Hc').
(cis-4a-transoid-4a,5-cis-5)-11,11-dimethyl-1,2':4,2"-dipyridyl-4a,5,10,10a-tetrahydro-5,10-methanobenzo[g]phthalazine (39) and (transoid-4a, 5-cis-5)-11,11-dimethyl-l,2':4, 2"-dipyridyl-2,4a,5,10-tetrahydro-5,10-methanobenzo[g]phthalazine (40).
Similarly, treatment of 1,4-dihydro-9,9-dimethyl-1,4-methanonaphthalene (25) with (2) gave a mixture of (39) and (40) The mixture was not purified, and characterised only by 1H n.m.r.spectroscopy. 1H n.m.r. spectrum (80 MHz, CDC13): d 0.65, 0.81, 1.38,1.42 (4x s, methyl protons), 3.52 (dd Ja b= 2.0 Hz, Ja' b= 2.0 Hz, Hb), 3.88 (d Ja' b'= 3.6 Hz, Hb'). 3.92 (s, Hd'), 4.16 (dd Ja' b'= 3 6 Hz, J a' d'= 1.4 Hz, Ha'), 4.43 (dd Ja,b= 2.0 Hz, Ja,b'= 2-0 Hz, H a), 6.0-9.0 (m, aromatic protons), 8.62 (s, exchanged with DO-/ D2O, Hc').
(transoid-4'a, 5'-cis-5')-2-Acetyl-1',2":4',2"'-dipyridyl2',4'a,5',10'-tetrahydrospiro [cyclopropane-l, 11'-[5,10]methanobenzo[g]phthalazine] (38).
A solution of the mixture of (31) and (34) (150 mg, 0.4 mmol) and pyridine (4 drops) in benzene (5 ml) was heated to reflux and treated, slowly, with a solution of acetyl chloride (200 ml, 2.4 mmol) in benzene (2 ml). The resulting mixture was stirred and heated under reflux overnight. The mixture was allowed to cool, ether (50 ml) added, and washed with brine (2 x 50 ml). The resulting solution was dried and freed of solvent to yield the crude product. Recrystallization from chloroform / n-hexane gave the product as colourless prisms (137 mg, 82%), m.p. 221-222 oC. U.v. lmax (e) 243 (16000), 273 (13700), 280 infl. (13300), 322 (2970) nm. I.r. nmax 1685s, 1590s, 1570s, 1470s, 1465s, 1440s, 1380s, 1360s, 1405s, 1400s, 1370m, 1350m, 1310w, 1180s, 1155s, 1130w, 1115m, lO90w, 1080m, 1045m, 1020m, 995s, 990s, 950w, 940m, 890m, 880w, 805w, 785s, 760s, 750s, 735s, 705s, 680m, 665s, 650s, 640s, 625m cm.-1 1H n.m.r. spectrum (80 MHz, CDCl3): d 0.4-1.0 (4H, m, cyclopropane protons), 2.32 (3H, s, CH3), 3.64 (lH, d Ja, c= 1 4 Hz, Hc), 4.00 (1H, dd Ja,b=3.8 Hz, Ja, c= 1.4 Hz, Ha), 4.12 (lH, d, Ja, b= 3 8 Hz, Hb), 6.05 (lH, dd J = 1.7, 6.1 Hz, Hd), 6.3-8.8 (11H, m, aromatic protons). Mass spectrum: m/z 419 (12%), 418 (M+, 32) (Found: M+, 418.1794. C27H22N40 requires 418.1794), 417 (12), 376 (33), 375 (56), 360 (18), 359 (47), 358 (12), 347 (15), 315 (26), 314 (100), 298 (33), 297 (13), 281 (11), 272 (27), 271 (34), 256 (11), 255 (18), 254 (11), 248 (27), 247 (52), 105 (14), 79 (10), 78 (23), 43 (30).
(cis-5')-5',10'-Dihydro-1', 2": 4', 2"'-dipyridyl-spiro[cyclopropane1,11'-[5,10]methanobenzo [g]phthalazine] (33)
A solution of the mixture of (31) and (35) (376 mg, 1.0 mmol), DDQ (250 mg, 1.1 mmol) and acetic acid (5 drops) in dioxan (10 ml) was stirred for 1 h. The resulting mixture was filtered and freed of solvent under vacuum to yield the crude product. Purification by preparative tlc (silica / 1:2 ethyl acetate / n-hexane) afforded the product as buff coloured prisms (265 mg, 71%), m.p. 201-202 oC (Found: C, 80.2; H, 4.7; N, 14.8. C25Hl8N4 requires C, 80.2; H, 4.9; N, 15.0%). U.V. lmax (e) 288 (31700) nm. I.r.nmax 3100w, 3065m, 3045m, 3025m, 1940w, 1590s, 1575s, 1550m, 1475s, 1460s, 1440m, 1425s, 1380s, 1365s, 1285w, 1245s, 1240m, ll90m, 1165w, 1150m, 1135m, lll0s, 1085m, 1045w, 1020w, 1015m, 1005w, 995m, 990m, 985w, 975m, 950w, 920w, 910m, 885w, 855w, 800s, 780s, 765m, 755w, 745s, 735s, 710s, 670s, 665m, 655s, 625m, 610s cm.-1 1H n.m.r. spectrum (80 MHz, CDCl3): d 0.7 (4H, m, cyclopropane protons), 5.08 (2H, s, Hc), 6.99 (2H, dd Ja, b= 5.3 Hz, Ja', b= 3.2 Hz, Hb), 7.31 (2H, ddd Jd, e= 4.7 Hz, Je, f=7.6 Hz, Je, g= 1.3 Hz, He), 7.49 (2H, dd Ja, b=5.3 Hz, J a, b'=3.2 Hz, Ha), 7.83 (2H, ddd Jd, f= 1 9 Hz, Je, f= 7.6 Hz, Jf, g=7.8 Hz, Hf), 8.62 (2H, ddd Jd,g=1.2 Hz, Je, g=1.3 Hz, Jf, g= 7 8 Hz, Hg ), 8.79 (2H, ddd Jd, e=4.7 Hz, Jd,f=1 9 Hz, Jd, g=1.2 Hd). mass spectrum: m/z 375 (29%), 374 (M+, 100), 373 (44), 346 (21), 345 (18),296 (18), 269 (7), 142 (15), 141 (21).
(cis-5')-5', 10'-dihydro-1', 2'':4', 2"'dipyridylspiro[cyclopentane-1,11'[5,10]methanobenzo[g]phthalazine] (44)
Oxidation of a mixture of (42) and (43), as above, afforded the title compound (44) as buff-coloured prisms from chloroform / n-hexane (63%), m.p. 149-150 oC. (Found: C, 80.4; H, 5.6; N, 13.8. C27H22N4 requires C, 80.6; H, 5.5; N, 13.9%). U.v. lmax (e) 287 (29400), 338 infl. (640) nm. I.r. nmax 3060s, 3030s, 3010s, 1990w, 1950w, l910w, 1890w, 1870w, 1590s, 1580s, 1555s, 1480s, 1440s, 1430s, 1370s, 1340w, 1310w, 1290w, 1250s, 1200s, 1170w, 1150w, 1135s, lll0s, 1080s, 1045m, l0l0m, 995s, 970w, 955w, 935w, 895w, 880w, 800s, 775s, 740s, 720s, 665s, 645s, 620s, 615s cm.-1 1H n.m.r. spectrum (80 MHz, CDCl3): d 1.53 (8H, m, cyclopentane protons), 5.29 (2H, s, Hc), 6.95 (2H, dd Ja b=5.2 Hz, Ja', b=3 1 Hz,Hb), 7 28 (2H, ddd Jd, e=4.8 Hz, Je,f=7-6 Hz, Je,g=1.4 Hz, He) 7.44 (2H, dd Ja b=5.2 Hz, Ja b'= 3 1 Hz, Ha), 7.80 (2H, ddd Jd, f=1 9 Hz, Je, f=7.6 Hz, Jf, g= 7.8, Hf), 8.60 (2H, ddd Jd, g=1.2 Hz, Je, g=1.4 Hz, Jf, g=7.8 Hz, Hg) 8.81 (2H, ddd Jd, e=4.8 Hz, Jd, f=1 9 Hz, Jd, g=1.9 Hz, Hd). Mass spectrum: m/z 403 (31), 402 (M+, 100), 401 (23), 385 (26), 384 (10), 375 (17), 374 (60), 373 (17), 335 (16), 333 (17), 332 (7), 325 (11), 324 (44), 321 (12), 320 (42), 307 (16), 296 (20), 295 (58), 256 (11), 186 (11), 141 (11), 79 (10), 78 (39).
(cis-5)-5,10-dihydro-11,11-dimethyl-1,2':4,2"-dipyridyl-5,lO-methanobenzo[g]-phthalazine (41).
Oxidation of a mixture of (39) and (40) afforded the title compound (41) as buff-coloured prisms from chloroform / n-hexane (68%), m.p. 199-201 oC. (Found: C, 79.4; H, 5.4; N, 15.0. C25H20N4 requires C, 79.8; H, 5.4; N, 14.9%). U.v.lmax (e) 287 (22000) nm. I.r. nmax 3060m, 3040m, 1590s, 1580s, 1550s, 1475s, 1455s, 1430s, 1365s, 1260m, 1250s, 1225m, 1200w, 1180w, 1165w, 1145w, 1130m, ll00s, 1080m, 1040m, l0l0m, 990m, 970w, 930w, 890w, 885w, 880w, 800s, 780w, 770m, 760m, 740s, 715s, 680m, 655s, 630s, 625m cm-l. 1H n.m.r. spectrum (80 MHz, CDCl3): d 1.03 (3H, s, CH3) , 1.05 (3H, s, CH3), 5.17 (2H, s, Hc), 6.96 (2H, dd Ja, b=5.2 Hz, Ja', b=3 1 Hz, Hb), 7.37 (2H, ddd Jd, e=4.7 Hz, Je,f=7.6 Hz, Je, g=1.2 Hz, He), 7.42 (2H, dd Ja, b=5.2 Hz, Ja,b'=3 1 Hz, Ha), 7.86 (2H, ddd Jd, f= 1.8 Hz, Je, f=7.6 Hz, Jf, g= 7.8 Hz, Hf), 8.59 (2H, ddd Jd, g=1 1 Hz, Je, g= 1.2 Hz, Jf, g= 7 8 Hz, Hg), 8.82 (2H, ddd Jd, e=4-7 Hz, Jd, f=18 Hz, Jd, q=1.1 Hz, Hd). Mass spectrum: m/z 377 (20), 376 (M-, 69), 363 (29), 362 (100), 335 (23), 333 (10), 321 (15), 298 (17), 78 (21).
References
1. a) Warrener, R. N. in "Advances in Theoretically Interesting Molecules" Ed R. P. Thummel, JAI Press Inc, Greenwich, Connecticut, 1992, 2, 143-205. b) Warrener, R. N.; Pitt, I. G.; Weerasuria, K. D. V.; Russell, R. A. Aust. J. Chem. 1992, 45, 155-178. c) Warrener, R. N.; Russell, R. A.; Solomon, R.; Pitt, I. G.; Butler, D. N. Tetrahedron Lett. 1987, 28, 6503-6506.
2. a) Sauer, J.; Heinrichs, G. Tetrahedron Lett. 1966, 4979-4984. b) Barlow, M. G.; Haszeldine, R. N.; Pickett, J. A. J. Chem. Soc., Perkin Trans. 1 1978, 378-380.
3. Wilson, W. S.; Warrener, R. N. J. Chem. Soc., Chem. Commun. 1972, 211-212.
4. a) Warrener, R. N. J. Amer. Chem. Soc. 1971 93, 2346-2348. b) Russell, R. A.; Longmore, R. W.; Warrener, R. N.; Weersuria, K. D. V. Aust. J. Chem. 1991, 44, 1341-1345.
5. a) Priestly, G. M.; Warrener, R. N. Tetrahedron Lett. 1972, 4295-4298. b) Sun, G.; Butler, D. N.; Warrener, R. N.; Margetic, D.; Malpass, J. R. 062, Electronic Conference on Heterocyclic Chemistry '98", 1998, H.S. Rzepa and O. Kappe (Eds), Imperial College Press, ISBN 981-02-3594-1. (http://www.ch.ic.ac.uk/ectoc/echet98/pub/062/index.htm). c) Malpass, J. R.; Sun, G.; Fawcett, J.; Warrener, R. N. Tetrahedron Lett. 1998, 39, 3083-3086.
6. a) Watson, P. L.; Warrener, R. N., Aust. J. Chem., 1973, 26, 1725-1750. b) Warrener, R. N.; Russell, R. A.; Collin, G. J., Tetrahedron Lett. 1978, 4447-4450 c) Warrener, R. N.; Harrison, P. A.; Russell, R. A. Article 081, Electronic Conference on Heterocyclic Chemistry '98", 1998, H.S. Rzepa and O. Kappe (Eds), Imperial College Press, ISBN 981-02-3594-1. (http://www.ch.ic.ac.uk/ectoc/echet98/pub/081/index.htm).
7. a) Warrener, R. N., Schultz, A. C.; Butler, D. N.; Wang, S.; Mahadevan, I. B.; Russell, R. A. Chem. Commun.1997, 1023-1024. b) Schultz, A. C.; Kelso, L. S.; Johnston, M. R.; Warrener, R. N.; Keene, F. R. Inorg. Chem. 1999, 38, 4906. c) Warrener, R. N.; Schultz, A. C.; Johnston, M. R.; Gunter, M. J. J. Org. Chem., 1999, 64, 4218. d) Warrener, R. N.; Margetic, D.; Amarasekara, A. S.; Russell, R. A.; Org. Lett., 1999, 1, 203. e) Warrener, R. N.; Butler, D. N.; Russell, R. A. Synlett 1998, 556. f) Warrener, R. N.; Margetic, D.; Amarasekara, A. S.; Foley, P.; Butler, D. N.; Russell, R. A. Tetrahedron Lett., 1999, 40, 4111-4114. g) Warrener, R. N.; Margetic, D.; Russell, R. A. Article 014, Electronic Conference on Heterocyclic Chemistry '98", 1998, H. S. Rzepa and O. Kappe (Eds), Imperial College Press (review). h) Warrener, R. N.; Butler, D. N.; Russell, R. A. Synlett,1998, 566 (review). i) Butler, D. N.; Malpass, J. R.; Margetic, D.; Russell, R. A.; Sun, G.; Warrener, R. N. Synlett,1998, 588. j) Sun, G.; Butler, D. N.; Warrener, R. N.; Margetic, D.; Malpass, J. R. Article 062, Electronic Conference on Heterocyclic Chemistry '98",1998, H. S. Rzepa and O. Kappe (Eds), Imperial College Press. (http://www.ch.ic.ac.uk/ectoc/echet98/pub/062/index.htm) k) Butler, D. N.; Hammond, M. L. A.; Johnston, M. J.; Sun, G.; Malpass, J. R.; Fawcett, J.; Warrener, R. N. Org. Lett. 2000, 2, 721-724. l) Malpass, J. R.; Butler, D. N.; Johnston, M. J.; Hammond, M. L. A.; Warrener, R. N. Org. Lett., 2000, 2, 725-728. m) Warrener, R. N.; Butler, D. N.;. Margetic, D; Pfeffer,F. M.; Russell, R.A. Tetrahedron Lett. 2000, 41, 4671-4675.
8. For alternative types of 'lego' building block techniques, see Ashton, P. R.; Girreser, U.; Giuffrida, D.; Kohnke, F. H.; Mathias, J. P.; Raymo, F. M.; Slawin, A. M. Z.; Stoddart, J. F.; Williams, D. J. J. Am. Chem. Soc. 1993, 115, 5422-5429. b) Pabst, G. R.; Sauer, J. Tetrahedron Lett. 1998, 39, 8817-8820.
9. Eilbracht, P.; Dahler, P. Annalen, 1979, 1890.
10. Miranof, V. A. et al. Tetrahedron Lett. 1969, 3347.
11. a) Avram, M.; Dinulescu, I.; Marcia, E.; Nenitzescu, C.D., Chem. Ber., 1962, 95, 2248. b) Satish, S.; Mitra, A.; George, M.V. Tetrahedron 1979, 35, 277.
12. a) Warrener, R. N.; Elsey, G. M.; Sankar, I. V.; Butler, D. N.; Pekos, P.; Kennard, C. H. L. Tetrahedron Lett. 1994, 35, 6745-6748. b) Paddon-Row, M. N.; Patney, H. K.; Warrener, R. N., J. Org. Chem. 1979, 44, 3908-3917.
13. Jackman, L.M. and Sternhell, S., "Applications of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry", 2nd Edit., pp.280-301. (Pergamon Press: Braunschweig 1969), p 211.
All comments on this poster should be sent by e-mail to (mailto:[email protected]) [email protected] with A0072 as the message subject of your e-mail.