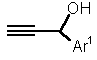[A0074]
A Coupling-Isomerization Sequence As An Entry To A Novel Three Component One-Pot Synthesis of 1,5-Benzoheteroazepines
Thomas J. J. Müller*, and Roland U. Braun
Department Chemie, Ludwig-Maximilians-Universitat München, Butenandtstr.
5-13 (Haus F), D-81377 München, Germany
E-mail: [email protected]
Received: 4 August 2000 / Uploaded: 5 August 2000
1,4- and 1,5-Benzodiazepines (1 and 2) constitute an important class of psychopharmaca [1], in particular, as tranquilizers and also as potent virucides and non-nucleoside inhibitors of HIV-1 reverse transcriptase [1,2]. Besides these only nitrogen containing benzoannealed seven-membered heterocycles their oxaza and thiaza analogues 3 have become increasingly interesting since 1,5 benzothiazepines show anti-fungal, anti-bacterial [3], anti-feedant [4], anti-inflammatory, analgesic [5], and anti-convulsant [6] activity.

Chart 1.
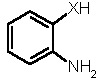
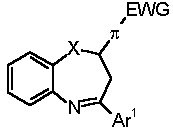
Retrosynthetically, 1,5-benzoheteroazepines are synthesized by cyclocondensation of the corresponding 2-substituted anilines with suitable enones or 1,3-dicarbonyl compounds (and synthetic equivalents) [3-6,7]. However, the enones and, in particular, chalcones (i.e. 1,3-diaryl enones) are usually prepared by an aldol condensation and have to be isolated and purified prior to the cyclization step. Therefore, we set out to develop a novel synthesis of 1,5-benzoheteroazepines, preferentially in a straightforward highly convergent manner, that also can be conducted in the sense of a one-pot process. Here, we wish to communicate a facile one-pot synthesis of 2,4-di(hetero)aryl substituted 2,3-dihydro 1,5-benzodiazepines, -oxazepines, and thiazepines (3, R1 = (het)aryl, R2 = aryl) based upon a coupling-isomerization sequence with a subsequent cyclocondensation with 2-amino, 2-hydroxy, or 2-mercapto anilines.
Scheme 1. One-pot pyrazoline and pyrimidine synthesis based upon a coupling-isomerization sequence.

Recently, we found that palladium/copper catalyzed cross-coupling reactions of electron poor halogen substituted p-systems and 1-aryl prop-2-yn-1-ols do not furnish the expected propargyl alcohols but the isomeric enone components [8]. Mechanistically, this isomerization occurring after the cross-coupling reaction is purely base catalyzed and opens a new access to electron deficient propenones. With this powerful tool for the construction of chalcones (1,3-diaryl propenones) in hand and considering the mild reaction conditions for the Sonogashira coupling reaction we have developed novel one-pot pyrazoline [8] and pyrimidine [9] syntheses (Scheme 1).
Since cyclocondensations of 2-heteroatom substituted anilines with chalcones (1,3-diaryl propenones) give 1,5-benzoheteroazepines [2-6], retrosynthetically, an extension of the coupling-isomerization-based methodology to a one-pot synthesis of 1,5-benzoheteroazepines can be easily envisioned. Upon cyclocondensing ortho-phenylene diamine, 2-amino phenol, or 2-amino thiophenol as suitable 1,4-dinucleophilic components with the initially formed enone functionality the benzoannealed seven-membered heterocycles are to be readily formed (Scheme 2). In particular, the mild reaction conditions of Sonogashira couplings [10] not only allow the presence of sensitive functional groups without tedious protection and deprotection steps but are also advantageous for base-mediated processes such as cyclocondensations. In addition, this strategy could also be extended to a combinatorial approach to 1,5-benzoheteroazepines (3).
Scheme 2. Retrosynthetic concept for a three component 1,5-benzoheteroazepine synthesis.

Thus, we have submitted p-iodo nitrobenzene (4a), 4-bromo pyridine (4b), or p-bromo benzonitrile (4c), aryl propynols 5 [11] and 2-heteroatom substituted anilines 6 to the reaction conditions of the Sonogashira coupling in boiling mixture of triethylamine and THF [12]. In all cases the isolated products were the beige to yellow 1,5-benzoheteroazepines 7 in 32-67 % yield (Table 1) [13]. As already shown for the one-pot synthesis of pyrazolines and pyrimidines the electron withdrawing nature of the (hetero)aryl halide 3 is crucial for the successful coupling-isomerization step [8,9].
The proton and carbon NMR spectroscopic data support the formation of the 1,5-benzoheteroazepine, in particular, in the 1H NMR spectra of 7 by the indicative appearance of the ABM-spin system with the characteristic geminal and vicinal coupling constants for the methylene group resonances (2J = 13.5 Hz, 3J = 4.4 Hz, 3J = 7.1 Hz) and the vicinal coupling constants for the methine resonances (3J = 4.4 Hz, 3J = 7.0 Hz). Furthermore, the structure of 7 was unambiguously supported by an X-ray crystal structure analysis (Figure 1) of compound 7a (Table 1, Entry 1).
Table 1. Three-component 1,5-Benzoheteroazepine Synthesis Based upon a Coupling-Isomerization-Cyclocondensation Sequencea.
 |
||||
| Entry | EWG-p-Hal 3 | Propargyl alcohol 5
|
2-Hetero aniline 6
|
1,5 Benzoheteroazepine 7 (Yield %)b
|
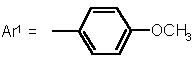
| 1 | 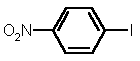 4a |
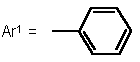 5a |
X = NH 6a |
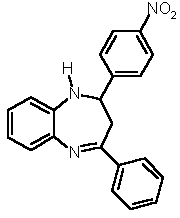 7a (52 %) 7a (52 %) |
| 2 | 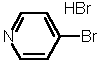 4b |
5a |
X = NH 6a |
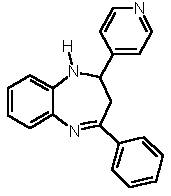 7b (53 %) 7b (53 %) |
| 3 | 4a |
 5b |
X = NH 6a |
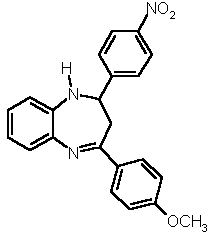 7c (42 %) 7c (42 %) |
| 4 | 4a |
5a |
X = O 6b |
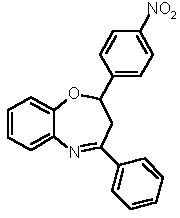 7d (35 %) 7d (35 %) |
| 5 | 4a |
 5b |
X = O 6b |
 7e (32 %) 7e (32 %) |
| 6 | 4a |
5a |
X = S 6c |
 7f (67 %) 7f (67 %) |
| 7 | 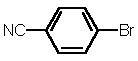 4c |
5a |
X = S 6c |
 7g (67 %) 7g (67 %) |

Figure 1
In conclusion, we could show that the mild reaction conditions of the coupling-isomerization sequence of electron poor (hetero)aryl halides with 1-aryl propargyl alcohols giving rise to chalcones can be extended to a one-pot three component synthesis of 2,4-di(hetero)aryl substituted 2,3-dihydro 1,5-benzodiazepines, -oxazepines, and thiazepines. Since 2-heteroatom substituted anilines can act as chelating ligands as well transition metal templated syntheses of 14-membered rings [14] for novel ligands in catalysis can be easily envisioned. Further studies directed to extend these one-pot heterocycle syntheses are currently underway.
Acknowledgments. The financial support of the Fonds der Chemischen Industrie, Deutsche Forschungs-gemeinschaft and the Dr.-Otto-Rohm Gedachtnisstiftung is gratefully acknowledged. The authors wishes to express their appreciation to Dr. K. Polborn for performing the X-ray structure analysis and to Prof. H. Mayr for his generous support.
References and Notes
[1] For reviews, see e.g., (a) Archer, G. A.; Sternbach, L. H. Chem. Rev. 1968, 68, 747. (b) Popp, F. D.; Noble, A. C. Adv. Heterocycl. Chem. 1967, 8, 21. (c) Sternbach, L. H. Angew. Chem. Int. Ed. Engl. 1971, 10, 34. (d) Vanderheyden, J. L.; Vanderheyden, J. E. J. Pharm. Belg. 1981, 36, 354. (e) Jones, G. R.; Singer, P. P. Adv. Anal. Toxicol. 1989, 2, 1. (f) Newkome, G. R. in Comprehensive Heterocyclic Chemistry II (Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., eds.), Pergamon Press, Oxford, New York, Toronto, Sydney, Paris, Frankfurt, 1996, Vol. 9, 181.
[2] For recent pharmacological studies, see e.g., (a) Breslin, H. J.; Kukla, M. J.; Kromis, T.; Cullis, H.; De Knaep, F.; Pauwels, R.; Andries, K.; De Clercq, E.; Janssen, M. A. C.; Janssen, P. A. J. Bioorg. Med. Chem. 1999, 7, 2427. (b) Smith, R. H. Jr.; Jorgensen, W. L.; Tirado-Rives, J.; Lamb, M. L.; Janssen, P. A. J.; Michejda, C. J.; Smith, M. B. K. J. Med. Chem. 1998, 41, 5272. (c) Lokensgard, J. R.; Chao, C. C.; Gekker, G.; Hu, S.; Peterson, P. K. Mol. Neurobiol. 1998, 18, 23. For synthetic studies, see e.g., (d) Parker, K. A.; Dermatakis, A. J. Org. Chem. 1997, 62, 4164. (e) Farese, A.; Peytou, V.; Condom, R.; Sinet, M.; Patino, N.; Kirn, A.; Moog, C.; Aubertin, A. M.; Guedj, R. Eur. J. Med. Chem. 1996, 31, 497. (f) Liu, J.; Dodd, R. H. J. Heterocycl. Chem. 1995, 32, 523. (g) Salaski, E. J. Tetrahedron Lett. 1995, 36, 1387.
[3] (a) Mane, R. A.; Ingle, D. B. Indian J. Chem., Sect. B 1982, 21B, 973. (b) Jadhav, K. P..; Ingle, D. B. Indian J. Chem., Sect. B 1983, 22B, 180. (c) Attia, A.; Abdel-Salam, O. I.; Abo-Ghalia, M. H.; Amr, A. E. Egypt. J. Chem. 1995, 38, 543.
[4] Reddy, R. J.; Ashok, D.; Sarma, P. N. Indian J. Chem., Sect. B 1993, 32B, 404.
[5] Satyanarayana, K.; Rao, M. N. A. Indian J. Pharm. Sci. 1993, 55, 230.
[6] DeSarro, G.; Chimirri, A.; DeSarro, A.; Gitto, R.; Grasso, S.; Zappala, M. Eur. J. Med. Chem. 1995, 30, 925.
[7] Lloyd, D.; McNab, H. Adv. Heterocycl. Chem. 1998, 71, 1.
[8] Müller, T. J. J.; Ansorge, M.; Aktah, D. Angew. Chem. Int. Ed. 2000, 39, 1253.
[9] Müller, T. J. J.; Braun, R.; Ansorge, M. Org. Lett. 2000, 2, 1967.
[10] (a) Takahashi, S.; Kuroyama, Y.; Sonogashira, K.; Hagihara, N. Synthesis 1980, 627. (b) Sonogashira, K. in Metal catalyzed Cross-coupling Reactions (Diederich, F., Stang, P. J., Eds.; Wiley-VCH, Weinheim, 1998, 203.
[11] The propynols 4 were synthesized according to Krause, N.; Seebach, D. Chem. Ber. 1987, 120, 1845.
[12] Typical procedure (7g, entry 7): To a magnetically
stirred solution of 0.25 g (1.00 mmol) 4-brome benzonitrile (4c), 22 mg (0.02 mmol)
of Pd(PPh3)2Cl2, and 2 mg (0.01 mmol) of CuI in degassed
mixture of 6 mL of THF and 3.5 mL of triethylamine under nitrogen a solution of 139 mg
(1.05 mmol) of 1-phenyl propyn-1-ol (5a) in 6 mL of THF was added dropwise at room
temperature over a period of 30 min. The mixture was heated to reflux temperature for 16
h. After cooling to room temperature 138 mg (1.10 mmol) of 2-amino thiophenol (6c)
was added and the reaction mixture was heated to reflux temp. for 8 h. After cooling the
solvents were removed in vacuo and the residue dissolved in dichloromethane and was
filtered through a short pad of silica gel. The solvents were evaporated in vacuo
and the residue was recrystallized from dichloromethane/pentane to give 228 mg (67 %) of
analytically pure 7g as yellow needles. Mp. 180-181 �C. 1H-NMR (CDCl3,
300 MHz): d 3.03 (t, J = 12.8, 1 H), 3.30 (dd, J
= 4.9 Hz, J = 12.9 Hz, , 1 H), 4.97 (dd, J = 7.7 Hz, J = 12.6 Hz, 1
H), 7.16 (dt, J = 7.5 Hz, J = 1.4 Hz, 1 H), 7.32 (dd, J = 7.8, J
= 1.9, 1 H), 7.41 (d, J = 8.3 Hz, 2 H), 7.61-7.46 (m, 7 H), 8.04 (dd, J =
7.6, J = 1.4, 2 H). 13C-NMR (CDCl3, 300 MHz): d 36.7 (CH2), 59.4 (CH), 111.3 (Cquat.), 118.3
(Cquat.), 121.6 (Cquat.), 125.2 (CH), 125.3 (CH), 126.6 (CH), 127.1
(CH), 128.6 (CH), 130.0 (CH), 131.0 (CH), 132.4 (CH), 134.7 (CH), 137.1 (Cquat.),
148.6 (Cquat.), 152.2 (Cquat.), 168.1 (Cquat.). MS
(70 eV, m/z (%)): 340 (M+, 8), 211 (M+ - NCC6H4CH=CH2,
100). IR (KBr): 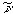 2229 cm-1,
1609, 1574, 1452. UV/Vis (CHCl3): l max
(e ) 244 nm (28700). Anal. Calcd. for C22H16N2S
(340.45): C, 77.62; H, 4.74; N, 8.23; S, 9.42. Found: C, 77.48; H, 4.76; N, 8.26; S, 9.52.
2229 cm-1,
1609, 1574, 1452. UV/Vis (CHCl3): l max
(e ) 244 nm (28700). Anal. Calcd. for C22H16N2S
(340.45): C, 77.62; H, 4.74; N, 8.23; S, 9.42. Found: C, 77.48; H, 4.76; N, 8.26; S, 9.52.
[13] All compounds have been characterized spectroscopically and by correct elemental analysis.
[14] Hideg, K.; Lloyd, D. J. Chem. Soc. C 1971, 20, 3441.
All comments on this poster should be sent by e-mail to (mailto:[email protected]) [email protected] with A0074 as the message subject of your e-mail.