[A0075]
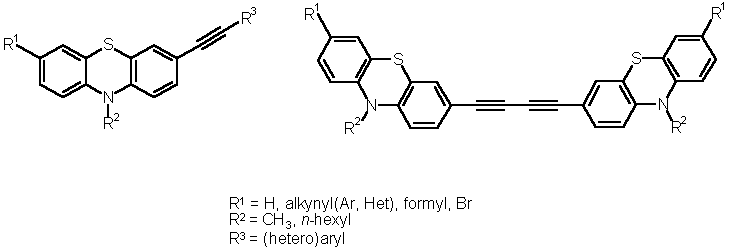
Syntheses of Functionalized Alkynylated Phenothiazines
Thomas J. J. Müller*, and Christa S. Kramer
Department Chemie, Ludwig-Maximilians-Universitat München, Butenandtstr.
5-13 (Haus F), D-81377 München, Germany
E-mail: [email protected]
Received: 4 August 2000 / Uploaded: 5 August 2000
Phenothiazines have proven to be a pharmaceutically important class of heterocycles [1], and due to their pharmacological efficacy they are applied as sedativa, tranquilizers, anti-epilectica, anti-tuberculotica, antipyretica, anti-tumor agents, bactericides and parasiticides [2]. Interestingly, phenothiazines are able to cleave DNA upon photochemical induction [3]. Fairly early, it was recognized that the low oxidation potential of this class of tricyclic nitrogen-sulfur heterocycles and their propensity to form stable radical cations play a key role in their physiological activities [4]. More recently, due to their reversible oxidation [1,5] phenothiazine derivatives have become attractive supramolecular [6] and material scientific [7] motifs.
Recently, we have found a straightforward access to 3-mono- and 3,7-dialkynylated phenothiazines 1 and 2 that are interesting building blocks for redox active oligomers [8]. Application of the Eglinton-coupling to monoalkynylated systems 1 (R1 = CH3, n-hexyl) gave rise to dumbbell-shaped butadiynyl-bridged diphenothiazinyl compounds 3. Both heterocyclic fragments are electronically coupled according to cyclic voltammetry, absorption and emission spectroscopy. Coupled redox systems integrated in conjugated chains could constitute a so far unknown class of redox addressable molecular wires, in particular, for a redox manipulation of single molecules with nanoscopic scanning techniques [9,10].

However, the incorporation of redox dumbbells like 3 into conjugated oligomers, symmetrically or unsymmetrically, demands flexible functionality for cross-coupling and/or condensation approaches. Here, we communicate the syntheses and structures of alkynylated and butadiynyl bridged phenothiazines with variable functional groups.
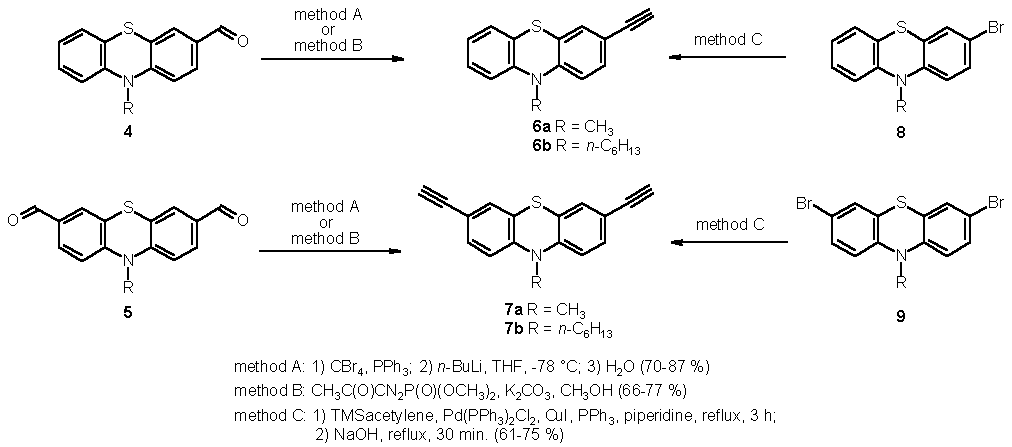
Synthetically, the exploitation of both aldehyde-alkyne transformations and cross-coupling methodologies opens flexible strategies to various functionalizations. Recently, we could show that phenothiazine 3-carbaldehydes 4 [11] and phenothiazine 3,7-biscarbaldehydes 5 [12], respectively, can be transformed to the alkynylated derivatives 6 and 7 according to the Corey-Fuchs protocol [13] in good yields (Scheme 1, method A).
Scheme 1
Alternatively, the fairly mild conditions of the Ohira-Bestmann transformation [14] of 4 and 5 to 6 [15] and 7 [15] opens a new access to alkynylated systems with broad functional group tolerance (method B). Additionally, we have transposed the Sonogashira ethynylation [16] to the mono- and dibrominated phenothiazines 8 [17] and 9 [18] to give the desired alkynylated derivatives after subsequent alkaline desilylation in one pot (method C). The X-ray crystal structure analysis of 7a [19] (Figure 1) clearly shows the expected butterfly-conformation [1] of the phenothiazine core with dihedral angles of 141.9 (C2-C1-S1-C12) and 140.0� (C5-C6-N1-C7). The bond lengths of the phenothiazinyl moiety and the triple bonds lie within the expected margins as well (C16-C17: 1.17 A; C13-C14: 1.16 A). Furthermore, the N-methyl group adopts a pseudoequatorial arrangement.
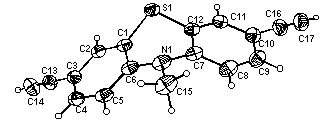
Figure 1
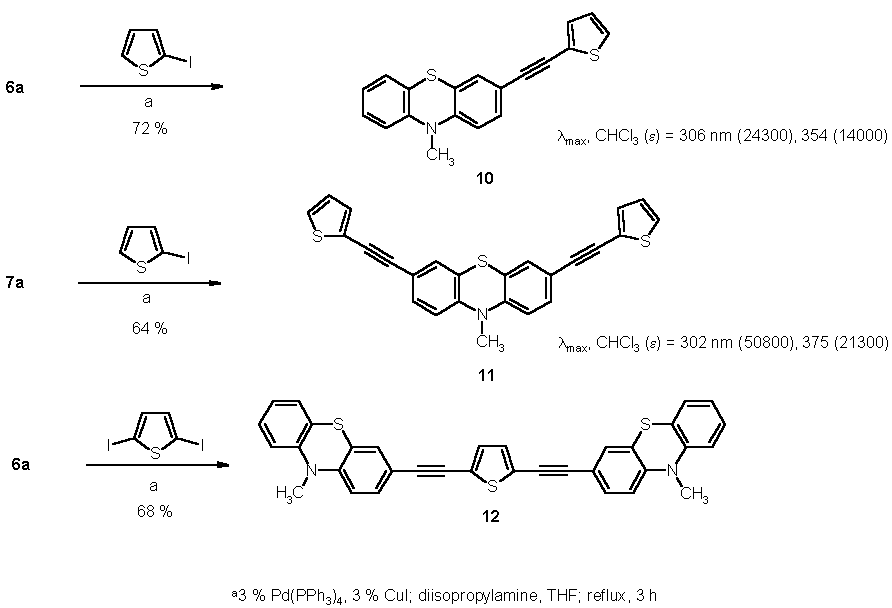
The mono- and diethynylated compounds 6 and 7 are suitable building blocks for alkynyl-bridged phenothiazine based redox systems and, thus, the Sonogashira coupling of 6a and 7a with 2-iodo thiophene and 2,5-diiodo thiophene, respectively, give rise to the formation of thienyl substituted (10 [20] and 11) and thienyl bridged (12) ethynyl phenothiazines (Scheme 2). [15]
Scheme 2
In the UV/Vis spectra of the thienyl ethynylated phenothiazines 10 and 11 the absorption bands at 306 (10) and 302 nm (11) arise from transitions of the phenylethynyl thiophene fragments as indicated by the doubling of molar extinction coefficients. However, the longest wavelength bands at 354 (10) and 375 nm (11) can be attributed to p-p* transitions within the extended p-system, i.e. including the conjugation through the nitrogen atom.
Finally, an entry to several functionalized alkynylated phenothiazines could be disclosed by bromination of the phenothiazine 3-carbaldehydes 4 in acetic acid to give 7-bromo phenothiazine 3-carbaldehydes 13 in good yields (Scheme 3). [21]
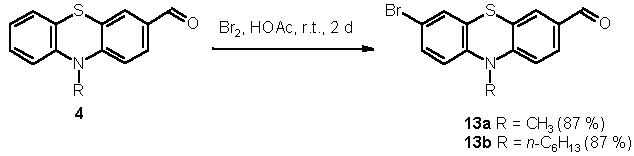
Scheme 3
With these unsymmetrically functionalized phenothiazines in hand now a selective functionalization of the bromo- or the formyl moiety could be successfully performed. Thus, the Ohira-Bestmann reaction of 13a furnishes the bromo alkyne 14 (60 %) that could be oxidatively dimerized by the copper mediated Eglinton coupling [22] to give the dibromo diyne 15 in good yields (Scheme 4). [15]

Scheme 4
Likewise, the Sonogashira coupling of 13 with TMSacetylene or phenylacetylene [23] gives rise to the alkynylated aldehydes 16 in decent to excellent yields [24]. Finally, the Eglinton coupling of 16a and 16b leads to the formation of the diformyl diynes 17 [25] in good yields [15].
In conclusion, we could show that alkynylated bromo and alkynylated formyl phenothiazines are easily accessible upon applying the mild conditions of the Ohira-Bestmann formyl-alkyne transformation or the Sonogashira coupling to the novel bromo formyl phenothiazine building block 13. Thus, the novel functionalized redox dumbbells 15 and 17 can be used as suitable starting materials for further synthetic elaboration towards molecular wires via cross-coupling and/or condensation strategies. Further studies directed towards polymer and oligomer syntheses with these novel ethynylated phenothiazines as well as the investigation of the electrochemical and photochemical behavior are currently underway.
Acknowledgments
The financial support of the Fonds der Chemischen Industrie, Deutsche Forschungsgemeinschaft (SFB 486) and the Dr.-Otto-Rohm Gedachtnisstiftung is gratefully acknowledged. The authors wishes to express their appreciation to Dr. K. Polborn for performing the X-ray structure analysis and to Prof. H. Mayr for his generous support.
References and Notes
[1] Sainsbury, M. in Comprehensive Heterocyclic Chemistry (Katritzky, A. R.; Rees, C. W.; eds.), Pergamon Press, Oxford, New York, Toronto, Sydney, Paris, Frankfurt, 1984, Vol. 3, 995.
[2] (a) Mietzsch, F. Angew. Chem. 1954, 66, 363. (b) Ionescu, M.; Mantsch, H. Adv. Heterocycl. Chem. 1967, 8, 83. (c) Bodea, C.; Silberg, I. Adv. Heterocycl. Chem. 1968, 9, 321. (d) Valzelli, L.; Garattini, S. in Prinicples of Psychopharmacology (Clark, W. G.; ed.), Academic Press, 1970, 255. (e) Okafor, C. O. Heterocycles 1977, 7, 391. (f) Eckstein, Z.; Urbanski, T. Adv. Heterocycl. Chem. 1978, 23, 1. (g) Szabo, J. Chem. Heterocycl. Compd. (USSR) (Engl. Trans.) 1979, 15, 291. (h) Albery, W. J.; Foulds, A. W.; Hall, K. J.; Hillman, A. R.; Edgell, R. G.; Orchard, A. F. Nature 1979, 282, 793.
[3] (a) Nishiwaki, E.; Nakagawa, H.; Takasaki, M.; Matsumoto, T.; Sakurai, H.; Shibuya, M. Heterocycles 1990, 31, 1763. (b) Decuyper, J.; Piette, J.; Lopez, M.; Merville, M. P.; Vorst, A. Biochem. Pharmacol. 1984, 33, 4025. (c) Motten, A. G.; Buettner, G. R.; Chignell, C. F. Photochem. Photobiol. 1985, 42, 9. (d) Fujita, H.; Matsuo, I. Chem. Biol. Interac. 1988, 66, 27.
[4] (a) Forrest, I.; Forrest, F. Biochim. Biophys. Acta 1958, 29, 441. (b) Iida, Y. Bull. Chem. Soc. Jpn. 1971, 44, 663. (c) Moutet, J.-C.; Reverdy, G. Nouv. J. Chim. 1983, 7, 105.
[5] McIntyre, R.; Gerischer, H. Ber. Bunsen Ges. Phys. Chem. 1984, 88, 963.
[6] (a) Duesing, R.; Tapolsky, G.; Meyer, T. J. J. Am. Chem. Soc. 1990, 112, 5378. (b) Jones, W. E. Jr.; Chen, P.; Meyer, T. J. J. Am. Chem. Soc. 1992, 114, 387. (c) Brun, A. M.; Harriman, A.; Heitz, V.; Sauvage, J.-P. J. Am. Chem. Soc. 1991, 113, 8657. (d) Burrows, H. D.; Kemp, T. J.; Welburn, M. J. J. Chem. Soc. Perkin Trans. 2 1973, 969. (e) Collin, J.-P.; Guillerez, S.; Sauvage, J.-P. J. Chem. Soc. Chem. Commun. 1989, 776.
[7] (a) Wheland, R. C.; Gillson, J. L. J. Am. Chem. Soc. 1976, 98, 3916. (b) Berges, P.; Kudnig, J.; Klar, G.; Sanchez-Martinez, E.; Diaz-Calleja, R. Synth. Met. 1992, 46, 207. (c) Knorr, A.; Daub, J. Angew. Chem. Int. Ed. Engl. 1995, 34, 2664. (d) Spreitzer, H.; Scholz, M.; Gescheidt, G.; Daub, J. Liebigs Ann. 1996, 2069. (e) Spreitzer, H.; Daub, J. Chem. Eur. J. 1996, 2, 1150.
[8] Müller, T. J. J. Tetrahedron Lett. 1999, 40, 6563.
[9] For applications of AFM and STM in chemistry, see e.g., (a) various authors, Chem. Rev. 1997, 97, issue 4. For applications in molecular electronics, see e.g., (b) Rabe, J. P. in An Introduction to Molecular Electronics (Petty, M. C.; Bryce, M. R., Bloor, D.; eds.), Oxford University Press, New York, 1995, 261. For nanoscale materials, see e.g., (c) various authors in Acc. Chem. Res. 1999, 32, issue 5.
[10] For conductance of single molecules under STM conditions, see e.g., (a) Bumm, L. A.; Arnold, J. J.; Cygan, M. T.; Dunbar, T. D.; Burgin, T. P.; Jones II, L.; Allara, D. L.; Tour, J. M.; Weiss, P. S. Science 1996, 271, 1705. (b) Davis, W. B.; Svec, W. A.; Ratner, M. A.; Wasielewski, M. R. Nature 1998, 396, 60. (c) Cygan, M. T.; Dunbar, T. D.; Arnold, J. J.; Bumm, L. A.; Shedlock, N. F.; Burgin, T. P.; Jones II, L.; Allara, D. L.; Tour, J. M.; Weiss, P. S. J. Am. Chem. Soc. 1998, 120, 2721. (d) Leatherman, G.; Durantini, E. N.; Gust, D.; Moore, T. A.; Moore, A. L.; Stone, S.; Zhou, Z.; Rez, P.; Liu, Y. Z.; Lindsay, S. M. J. Phys. Chem. B 1999, 103, 4006.
[11] Buu-Hoi, N.-P.; Hoan, N. J. Chem. Soc. 1951, 1834.
[12] Oelschlager, H.; Peters, H. J. Arch. Pharm. (Weinheim) 1987, 320, 379.
[13] Corey, E. J.; Fuchs, P. L. Tetrahedron Lett. 1972, 36, 3769.
[14] a) Ohira, S. Synth. Commun. 1989, 19, 561. b) Müller, S.; Liepold, B.; Roth, G. J.; Bestmann, H. J. Synlett 1996, 512.
[15] All new compounds have been characterized spectroscopically and by correct elemental analysis or HRMS (oils).
[16] (a) Takahashi, S.; Kuroyama, Y.; Sonogashira, K.; Hagihara, N. Synthesis 1980, 627. (b) Sonogashira, K. in Metal Catalyzed Cross Coupling Reactions, (Stang, P. J.; Diederich, F.; eds.), Weinheim, Wiley-VCH, 1998, 203.
[17] Cymerman-Craig, J.; Rogers, W. P.; Warwick, G. P. Aust. J. Chem. 1955, 8, 252.
[18] Bodea, C.; Terdic, M. Acad. Rep. Pop. Rom. 1962, 13, 81. Chem. Abstr. 1963, 59, 11477h.
[19] The X-ray crystal structure data will be published elsewhere.
[20] Synthesis of 10: To a degassed solution of 250 mg
(1.05 mmol) of 6a in 10 mL of dry diisopropylamine and 3 mL of THF were
successively added 211 mg (1.03 mmol) of 2-iodo thiophene, 35 mg (0.03 mmol) of Pd(PPh3)4,
and 6 mg (0.03 mmol) of CuI. The reaction mixture was heated to reflux temperature under
nitrogen for 3 h. After cooling to room temp. the residue was chromatographed on silica
gel (diethyl ether/pentane 1 : 4) to give 239 mg (72 %) of 10 as a light yellow
solid. Mp. 141 �C . 1H-NMR (CDCl3, 300 MHz): d
3.35 (s, 3 H, CH3), 6.72 (d, J = 8.4 Hz, 1 H), 6.79 (d, J = 8.1
Hz, 1 H), 6.93 (mc, 1 H), 6.98 (mc, 1 H), 7.10-7.19 (m, 2 H),
7.22-7.31 (m, 4 H). 13C-NMR (CDCl3, 75 MHz): d
35.4 (CH3), 82.5 (Cquat.), 92.5 (Cquat.), 113.8 (CH),
114.3 (CH), 116.8 (Cquat.), 122.8 (Cquat.), 122.8 (CH), 123.5 (Cquat.),
126.9 (CH), 127.0 (CH), 127.2 (CH), 127.5 (CH), 129.7 (CH), 130.8 (CH), 131.5 (CH), 145.1
(Cquat.), 145.9 (Cquat.). MS (70 eV, m/z (%)): 319 (M+, 100), 304 (74). IR (KBr):  1598 cm-1, 1574, 1519, 1461, 1442, 1328, 1261, 852,
805, 750, 705, 607. UV/Vis (CHCl3): l max
(e ) 267 nm (26100), 306 (24300), 354 (14000). Anal. Calcd. for
C19H13NS2 (319.4): C, 71.44; H, 4.10; N, 4.38; S, 20.07.
Found: C, 71.20; H, 4.22; N, 4.26; S, 19.87.
1598 cm-1, 1574, 1519, 1461, 1442, 1328, 1261, 852,
805, 750, 705, 607. UV/Vis (CHCl3): l max
(e ) 267 nm (26100), 306 (24300), 354 (14000). Anal. Calcd. for
C19H13NS2 (319.4): C, 71.44; H, 4.10; N, 4.38; S, 20.07.
Found: C, 71.20; H, 4.22; N, 4.26; S, 19.87.
[21] Synthesis of 13b: To a solution of 7.94 g (25.5
mmol) of 4 (R = n-hexyl) in 30 mL of glacial acetic acid was dropwise added
a solution of 1.30 mL (25.5 mmol) of bromine in 10 mL of glacial acetic acid. The redbrown
mixture was stirred at room temp. for 2 d. After addition of 300 mL of water and 600 mL of
diethyl ether the organic layer was dried with MgSO4. The solvents were removed
in vacuo and the residue was chromatographed on silica gel (diethyl ether/pentane 1 : 3)
to give 8.66 g (87 %) of 13b as a vicous darkbrown oil. 1H-NMR (CDCl3,
300 MHz): d 0.86 (t, J = 6.6 Hz, 3 H), 1.28-1.31 (m, 4
H), 1.41 (mc, 2 H), 1.77 (mc, 2 H), 3.82 (t, J = 7.2 Hz, 2
H), 6.69 (d, J = 8.6 Hz, 1 H), 6.87 (d, J = 8.4 Hz, 1 H), 7.19 (d, J =
2.2 Hz, 1 H), 7.23 (dd, J = 8.5 Hz, J = 2.2 Hz, 1 H), 7.54 (d, J =
1.8 Hz, 1 H), 7.62 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 9.78 (s, 1 H). 13C-NMR
(CDCl3, 75 MHz): d 13.9 (CH3), 22.5 (CH2),
26.4 (CH2), 26.5 (CH2), 31.3 (CH2), 48.0 (CH2),
114.9 (CH), 117.0 (CH), 115.7 (Cquat.), 124.3 (Cquat.), 126.0 (Cquat.),
128.3 (CH), 129.7 (CH), 130.2 (CH), 130.2 (CH), 131.2 (Cquat.), 142.6 (Cquat.),
150.2 (Cquat.), 189.8 (CH). MS (70 eV, m/z (%)): 391
(M+, 81Br, 100), 389 (M+, 79Br, 96). IR (KBr):
1688 cm-1, 1594, 1462, 1198.
UV/Vis (CHCl3): l max (e ) 246 nm (17200), 277 (20000), 385 (5600). Anal. Calcd. for C19H20NSOBr
(390.3): C, 58.46; H, 5.16; N, 3.59; S, 8.21; Br, 20.47. Found: C, 58.28; H, 5.23; N,
3.57; S, 8.02; Br, 20.40.
[22] a) Cadiot, P.; Chodkiewicz, W. in Chemistry of Acetylenes (Viehe, H. G.; ed.), Marcel Dekker, New York, 1969, 597. b) Scott, L. T.; Cooney, M. J. in Modern Acetylene Chemistry, (Stang, P. J.; Diederich, F.; eds.), VCH Weinheim, 1995, 321. c) Brandsma, L. Preparative Acetylene Chemistry, 2nd edition, Elsevier, Amsterdam, Oxford, New York, Tokyo, 1988, 212.
[23] Using the improved variation by Hundertmark, T.; Littke, A. F.; Buchwald, S. L.; Fu, G. C. Org. Lett. 2000, 2, 1729.
[24] Synthesis of 16c: To a solution of 11 mg (0.03 mmol) of
Pd(PhCN)2Cl2, 4 mg (0.02 mmol) of CuI and 0.24 mL (0.06 mmol) of a
0.25 M solution of PtBu3 in dioxane under nitrogen was
added 1 mL of dry dioxane to form a brown suspension. To this suspension were added 320 mg
(1.00 mmol) of 13a, 122 mg (1.20 mmol) of phenylacetylene, and a solution of 1.70
mL (1.20 mmol) of dry diisopropylamine in 8 mL of dioxane. The reaction mixture was
stirred for 2 d at room temp. After addition of 10 mL of ethyl acetate the mixture was
filtered through a short plug of silica gel. The solvents were removed from the yellow
filtrate in vacuo and the residue was chromatographed on silica gel (diethyl ether/pentane
1 : 1) to give 324 mg (95 %) of 16c as a voluminous bright yellow solid. Mp. 135
�C. 1H-NMR (CDCl3, 300 MHz): d 3.40 (s,
3 H,), 6.75 (d, J = 8.4 Hz, 1 H), 6.83 (d, J = 8.2 Hz, 1 H), 7.26 (d, J =
1.6 Hz, 1 H), 7.31-7.35 (m, 4 H), 7.48-7.51 (m, 2 H), 7.57 (d, J = 1.8 Hz, 1 H),
7.64 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 9.79 (s, 1 H). 13C-NMR
(CDCl3, 75 MHz): d 35.9 (CH3), 88.3 (Cquat.),
89.9 (Cquat.), 113.9 (CH), 114.5 (CH), 118.5 (Cquat.), 122.6 (Cquat.),
123.1 (Cquat.), 123.5 (Cquat.), 127.9 (CH), 128.2 (CH), 128.3 (CH),
129.9 (CH), 130.4 (CH), 131.2 (CH), 131.4 (Cquat.), 131.4 (CH), 143.9 (Cquat.),
150.3 (Cquat.), 189.9 (CH). MS (70 eV, m/z (%)): 341 (M+,
100). IR (KBr): 1687 cm-1,
1602, 1578, 1468. UV/Vis (CHCl3): l max
(e ) 295 nm (49000), 395 (11000). Anal. Calcd. for C22H15NSO
(341.4): C, 77.39; H, 4.43; N, 4.10; S, 9.39. Found: C, 77.06; H, 4.43; N, 4.03; S, 9.37.
[25] Synthesis of 17a: To a solution of 369 mg (1.39 mmol) of 16a
in 6 mL of methanol was added a solution of 379 mg (1.90 mmol) of copper(II) acetate
monohydrate in a mixture of 2 mL of methanol and 6 mL of pyridine. This reaction mixture
was heated to reflux temp. for 4 h. After cooling to room temp. the precipitated solid was
collected by suction and washed with methanol to give 308 mg (84 %) of a bright yellow
powder. T>250 �C (dec.). 1H-NMR (CDCl3, 300 MHz): d 3.44 (s, 6 H), 6.77 (d, J = 8.4 Hz, 2 H), 6.88 (d, J
= 8.3 Hz, 2 H), 7.32 (mc, 4 H), 7.60 (s, 2 H), 7.67 (d, J = 8.3 Hz, 2
H), 9.82 (s, 2H). MS (70 eV, m/z (%)): 528 (M+, 100), 513 (M+
- CH3, 22), 498 (M+ - 2 CH3, 21). IR (KBr): 2136 cm-1, 1685, 1600, 1576, 1467.
UV/Vis (CHCl3): l max (e ) 292 nm (74500), 408 (33000). Anal. Calcd. for C32H20N2S2O2
(528.6): C, 72.70; H, 3.81; N, 5.30; S, 12.13. Found: C, 72.66; H, 3.81; N, 5.42; S,
11.82.
All comments on this poster should be sent by e-mail to (mailto:[email protected]) [email protected] with A0075 as the message subject of your e-mail.