
[A0081]
Enantioselective Electrophilic Fluorination : Two Approaches Using Cinchona Alkaloids.
Dominique Cahard,* Christophe Audouard, Jérôme Baudoux, Barbara Mohar and Jean-Christophe Plaquevent
Université de Rouen, UPRES-A 6014 de l'IRCOF (Institut de
Recherche en Chimie Organique Fine), Rue Tesnière, 76821 Mont Saint Aignan Cedex, France.
E-mail: [email protected]
Received: 9 August 2000 / Uploaded: 15 August
Abstract : Naturally-occuring cinchona alkaloids were exploited in two different approaches for enantioselective fluorination. Firstly, the phase transfer catalysis involving cinchona-alkaloid-derived quaternary ammonium salts was investigated. Poor results were obtained (7% ee, 60% yield) in the fluorination of N-phthaloylphenylglycine ethyl ester. In an alternative method we describe the first ever enantiopure N-fluoro quaternary ammonium salts of cinchona alkaloids as enantioselective fluorinating agents. A one step transfer-fluorination on the alkaloids gave the [N-F]+ reagents. This new generation of fluorinating agents exhibit asymmetric induction ranging from 56 to 92% ee in the fluorination of enolates and silyl enol ethers.
Keywords: asymmetric synthesis, cinchona alkaloids, phase transfer catalysis, electrophilic fluorination.
In the continuing search for improved drugs and agrochemicals, fluorine compounds are known to exert a unique and profound influence on biological activity and selectivity. Most of these compounds are achiral, fluoro aromatics. Occasionally, such fluoro compounds are chiral but the fluorine is rarely attached at a stereogenic center. The importance of chirality in pharmacologically and biologically active molecules associated with the astonishing properties of fluorine has lead to huge efforts in the asymmetric synthesis of chiral non racemic fluoroorganic molecules with fluorine at a stereogenic center.1 The auxiliary controlled asymmetric synthesis of a-fluoro carbonyl compounds has so far been superior to the reagent controlled processes. Therefore, it is a very challenging problem to develop efficient processes for enantioselective fluorination. Our contribution to the enantioselective electrophilic fluorination utilises the naturally-occuring cinchona alkaloids as the source of chirality. Cinchona alkaloids have a venerable history in the field of asymmetric synthesis owing to their firmly established ability to induce asymmetry and they are widely used in asymmetric processes both in homogeneous and heterogeneous reactions. Asymmetric- Michael additions, Sharpless dihydroxylation, and hydrogenation with modified Pt-catalysts, as well as the catalytic enantioselective alkylation under phase transfer catalysis are amongst typical examples.2 Cinchona alkaloids are also used as chiral resolving agents and for chromatographic separation of enantiomers.
In our first approach, we were interested to develop electrophilic fluorination using achiral fluorinating reagents in the presence of quaternary cinchonidinium salts3 Ia-c and cinchoninium salts IIa-c (Figure 1).
Figure 1 : Asymmetric phase-transfer catalysts derived from cinchona alkaloids.
As it is known that electrophilic fluorinating reagents (N-fluorobenzenesulfonimide (NFSi), SelectfluorTM, and N-fluoropyridinium triflate (PyF+Tf-)) are not stable under basic aqueous conditions, we decided to explore solid-liquid phase transfer catalysis using the organic-soluble reagent NFSi which has a half-life t1/2< 20 h in NaOH (3 equiv)/Bu4NBr (0.1 equiv)/CH2Cl2 at room temperature. We selected N-phthaloylphenylglycine ethyl ester as a model compound and examined different reaction conditions using various solvents (CH2Cl2 /toluene, dichloroethane (DCE), MeCN), bases (NaOH, KOH, KOH-K2CO3, CsOH) and phase transfer catalysts (Ia-c, IIa-c). The best ee noted was 5% with a chemical yield of 10% with Ia/NaOH in DCE at -20°C.4 We also tested milder fluorinating reagents such as TsNFiPr and TsNFtBu, but no product was formed in these cases. Curiously, TsNFtBu was found to be stable under solid-liquid phase transfer conditions in contrast with the other N-F compounds. In addition, Schwesinger bases as organic-soluble bases were studied.5 Although under these conditions t1/2< 5 min was found for the decomposition of NFSi, up to 80% of the fluorinated product was isolated when the fluorinating reagent was added slowly. The best ee noted was 7% with 60% yield, using Ia (Scheme 1). No product was formed with PyF+Tf-, TsNFiPr, or TsNFtBu. Interestingly, fluorination with NFSi in the presence of (-)-N-benzyl-N-methylephedrinium bromide gave 65% of the fluorinated product with 5% ee displaying the opposite stereoselectivity to that observed with Ia.
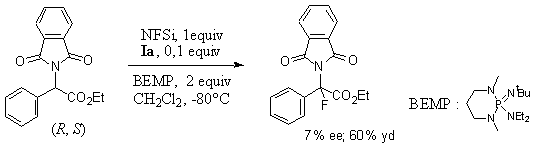
Scheme 1
Our second approach concerned the design, synthesis and evaluation of a novel class of enantioselective electrophilic fluorinating agents: N-fluoro ammonium salts of cinchona alkaloids F-CA-BF4. The pioneering work of E. Differding and R.W. Lang in 1988 lead to the development of the N-fluorocamphorsultam 1 as an enantioselective electrophilic fluorinating agent.6 F.A Davis et al, in 1993 and 1998 reported closely related structures 2.7 Recently, Y. Takeuchi et al designed the saccharin-based agents 38 as well as acyclic N-fluoro compounds 4 and 59 (Figure 2).

Figure 2 : Enantioselective fluorinating agents.
The fluorination of the enolate of 2-benzyl-1-tetralone by reagent 3 gave the highest enantiomeric excess of 88% in this series.8b The synthesis of these agents requires several steps, the ultimate step being the N-F bond formation, by means of either elemental fluorine F2 or FClO3. The unwelcome prospect of handling elemental fluorine as well as the wish to exploit the cinchona alkaloids lead us to proceed according to Bank's transfer-fluorination of the quinuclidine moiety with Selectfluor.10 We herein describe the synthesis, characterisation and applications of N-fluoro cinchona alkaloids which form a new class of enantioselective fluorinating agents.
At first Bank’s fluorine-transfer procedure was applied to an equimolar mixture of cinchonidine and Selectfluor in acetonitrile. Complete transfer was achieved within 30 minutes according to 19F NMR analysis of the reaction mixture. Acetonitrile was removed under reduced pressure and the resulting white solid was dissolved in acetone. Addition of a solution of H2SO4 in acetone caused the precipitation of 1-chloro-4-hydro-1,4-diazoniabicyclo [2.2.2] octane hydrogen sulphate tetrafluoroborate. To the filtrate was added diethyl ether that gave the precipitation of the N-fluoro cinchonidinium salt that was recrystallised in acetone to yield pure product. The structure of the N-fluoro ammonium salt was established by NMR spectroscopy and X-ray crystallography.11 It is a white solid, virtually non-hygroscopic, free flowing, crystalline and is high-melting (189°C). Cinchonine, quinine and quinidine were also sujected to the analogous fluorine-transfer procedure. Structures are depicted in figure 3.
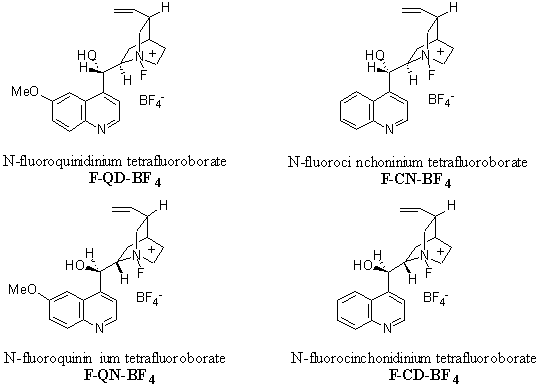
Figure 3 : N-fluoro cinchonium tetrafluoroborates.
In order to evaluate the ability of these reagents to promote enantioselective fluorination, we targetted a-fluoro carbonyl compounds via their enolates. All previously reported agents for enantioselective fluorination have been tested on the enolate of 2-methyl-1-tetralone as model substrate. The results of our evaluation on the same substrate allowed a direct comparison between the first generation of neutral N-F fluorinating agents and our new class of N-fluoro quaternary ammonium agents [N-F]+. Preliminary experiments demonstrated that sodium hydride in THF was a suitable base for quantitative conversion of 2-methyl-1-tetralone into its sodium enolate, however, the fluorination only yielded moderate amount (40-50%) of the expected 2-fluoro compound. A protonation of the enolate either by the free OH group of the alkaloid or by a molecule of water present in the crystalline structure, could explain the moderate yield of the fluorination. It is important to note that the protonation occured enantioselectively since an ee of 20% was measured on the recovered 2-methyl-1-tetralone. Indeed we circumvented this problem by using two equivalents of base and the fluorination became quantitative; alternatively, the hydroxyl function was protected. All four N-fluoro cinchonium salts were evaluated in the fluorination reaction, with F-CD-BF4 giving the highest enantioselectivity with the fluorinated stereocenter having the (S)-configuration7b (Scheme 2). The (R)-enantiomer was obtained when using the pseudoenantiomeric F-CN-BF4.
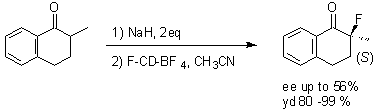
Scheme 2
Owing to the solubility characteristics of F-CA-BF4, the reactions were carried out either in a mixture THF/acetonitrile or under heterogeneous conditions with a suspension of F-CA-BF4 in THF with minor effect on the enantiomeric excesses. We are currently optimising both the fluorinating agents and the reaction conditions for a better enantioselectivity in the fluorination of enolates.
We also designed this new generation of N-fluoro ammonium salts for their stronger fluorinating power. Thus fluorination of enol derivatives such as silyl enol ethers can now be considered where such chiral neutral N-F fluorinating agents were discarded due to their low reactivity towards poor nucleophiles. The degree of asymmetric induction exhibited by the N-fluoro ammonium salts of cinchona alkaloids is strongly dependent on the reactions conditions with enantiomeric excesses up to 92% (Scheme 3).

Scheme 3
This approach involving silyl enol ether is more convenient than the fluorination of enolate since no base is required and also more promising on account of the higher asymmetric induction noted.
In summary electrophilic enantioselective fluorination under phase transfer catalysis gave poor results but the fluorination of enolates and better silyl enol ethers by means of a new N-fluoro ammonium salts of cinchona alkaloids affords quaternary a-fluoro carbonyl compounds in excellent yield and moderate to good enantiomeric excess.
Acknowledgment
This research is supported by RHODIA Organique Fine
References and notes
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with A0081 as the message subject of your e-mail.