[A0092]
Virtual Planar Chirality.
A New Approach to Catalyst and Ligand Design
Geraint Jones and Christopher J. Richards*
Department of Chemistry, Cardiff University, PO Box 912,
Cardiff, CF10 3TB, UK
E-mail: [email protected]
Received: 15 August 2000 / Uploaded: 20 August
Abstract: Attachment of a chiral imidazole or chiral oxazoline to either (h5-cyclopentadienyl)(h4-tetraphenylcyclobutadiene)cobalt or pentaphenylferrocene results in the heterocycle mimicking an environment of planar chirality. This chiral relay effect is demonstrated by both highly diastereoselective palladation and application of the oxazoline complexes as ligands in asymmetric catalysis.
Introduction
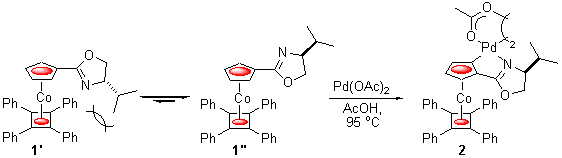
Planar chiral organometallics, and especially metallocenes such as ferrocene, have been extensively utilised as the basis of a wide variety of chiral ligands for application in asymmetric synthesis.1 As part of a programme investigating the synthesis of new planar chiral complexes, we recently reported that the oxazoline appended metallocene 1 undergoes highly diasteroselective metallation when heated with palladium acetate to give exclusively palladacycle 2 (Scheme 1).2 The outcome of this reaction was rationalised by the destabilisation of conjugated rotamer 1' over the alternative 1'' due to an unfavourable interaction in the former between the oxazoline isopropyl substituent and the tetraphenyl floor of the metallocene. As a consequence of these bulky moieties, the oxazoline group in 1 may be regarded as being in an environment of virtual planar chirality, i.e. as if the heterocycle were covalently bound to a second position of the cyclopentadienyl ring, as is actually the case in complex 2. This example of a chiral relay offers the potential to synthesise simple alternatives to planar chiral ligands that require only a single element of central chirality, and in this paper we report on our initial efforts towards this goal.
Scheme 1
Chiral Imidazole Appended Metallocene Catalysts
Recent interest in the development of non-enzymatic methods for enantioselective acyl transfer reactions has involved the use of both stoichiometric chiral acylating agents3 and nucleophilic catalysts employed for acyl transfer.4 Illustrative of the latter category is the pentaphenyl ferrocene derived structure 3 which has proved especially effective for the kinetic resolution by acylation of a range of secondary alcohols, and represents the best non-enzymatic method for carrying out this transformation.4b,d A consequence of the rotameric preference observed for 1 is the similarity of the oxazoline nitrogen environment to that of the pyridine nitrogen in the DMAP derivative. Although the oxazoline is unsuitable for use as a DMAP surrogate, imidazole and its derivatives have been extensively studied as nucleophilic catalysts for acyl transfer.5 We were intrigued by the possibility of attaching a 2-imidazoyl group to the metallocene, and examining the ability of a stereogenic centre attached to position 1 to control the stereochemical outcome of reactions occurring at the nucleophilic and basic nitrogen of this heterocycle.
Carboxylic acid 4, readily prepared from the reaction of diphenylacetylene and sodium carbomethoxycyclopentadienylide with CoCl(PPh3)3 followed by ester hydrolysis,2 was readily transformed into the amides 5a/b. Subsequent reaction of these yellow complexes with PCl5 in CH2Cl2 resulted in their conversion into intermediate red imidyl chlorides. These were immediately reacted with 2,2-dimethoxyethylamine to give, in both cases, amidines isolated by chromatography as a mixture of isomers. These were not separated but were instead converted with 6N HCl into imidazole appended metallocenes 6a/b in good overall yield (Scheme 2).
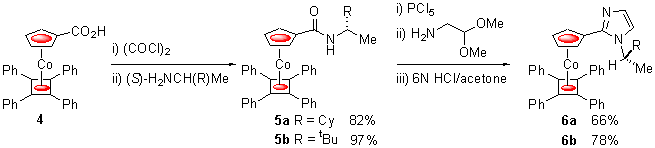
Scheme 2
The structure of 6a was confirmed by an X-ray crystal structure analysis (Figure 1). As anticipated the smallest substituent attached to the stereogenic centre, hydrogen, is pointing towards the bulky metallocene, with the smaller of the two remaining substituents oriented towards the tetraphenylcyclobutadiene floor. To aid accommodation of this methyl substituent one of the phenyls lies essentially perpendicular to the cyclobutadiene group to which it is attached. A consequence of this arrangement is the proximity of the active nitrogen N(2) to C(2), one of the two diastereotopic a -positions of the cyclopentadienyl ring, and the orientation of this nitrogen away from the bulk of the metallocene (C(2)-C(1)-C(10)-N(2) = 23.6 o).
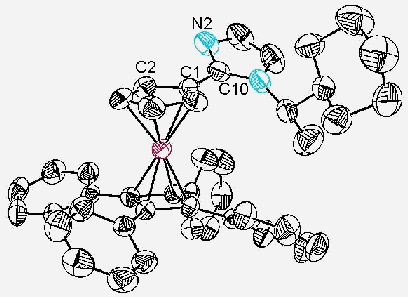
Figure 1. X-ray Crystal Structure of 6a
To examine if this conformation is maintained in solution, 1H NMR NOE difference spectra of 6a/b were recorded in CDCl3 at room temperature revealing, in both cases, the proximity of the stereogenic centre methines to predominantly one of the two diastereotopic a -positions of the cyclopentadienyl rings. The disparity in the population of rotamers is greater for the tert-butyl containing derivative 6b. Support for there being two, essentially conjugated, rotameric minima is provided by the proximity of the stereogenic centre methines to the ortho-hydrogens of the tetraphenycyclobutadiene moieties.
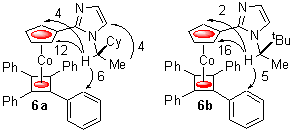
To test the kinetic accessibility of these rotamers, 6a/b were heated separately with Pd(OAc)2 for 30 minutes under the same conditions previously employed for the palladation of oxazoline 1. After cooling of the reaction mixtures to room temperature, both led to isolation by filtration of new palladated complexes 7a/b formed as single diastereoisomers, as revealed by the single sets of peaks observed in their 1H/13C NMR spectra (Scheme 3). Examination of the mother liquors revealed, in both cases, the presence of only the same diastereoisomer as isolated by filtration. The assumption that these reactions are under kinetic control was tested by heating 6a with Pd(OAc)2 for only two minutes prior to examination of the reaction mixture by NMR spectroscopy. Peaks corresponding to palladacycle 7a could clearly be identified and there was no evidence to suggest the presence of the alternative diastereoisomer. The configuration of the new element of planar chirality in these complexes was established as pR by an X-ray crystal structure analysis of 7b. Treatment of 7b with LiAlD4 in Et2O gave clean conversion to 8 with ~56% deuterium incorporation as determined by the reduction in the signal intensity for the peak at 5.45 ppm in the 1H NMR spectrum. This confirms the NOE assignments made for 6a/b, and the identity of 6' as the major rotamer present in solution, i.e. as also found in the solid state as determined by the X-ray structure analysis. That palladation occurred exclusively via this rotamer enables the imidazole to be regarded as in an environment of virtual planar chirality, with one face of the heterocycle blocked by the tetraphenylcyclobutadiene moiety of the adjoined complex. The alternative minor rotamer 6'' likely features nitrogen N(2) being tipped towards the tetraphenyl floor to minimise interaction of either the cyclohexyl or tert-butyl substituents with this bulky moiety, with the inaccessibility of the N(2) lone pair accounting for the inactivity of this rotamer.

Scheme 3
For a preliminary investigation into catalysis with 6a/b our attention focused on the alcoholysis of chlorophosphates for which an acceleration in half life time of 7 x 104 was reported for the ethanolysis of diethylchlorophosphate in the presence of one equivalent of N-methyl imidazole (NMI).6 This is reported to act as a nucleophilic catalyst in this reaction, and is a true catalyst as sub-stoichiometric quantities may be employed with NEt3 acting as a base. When dimethylchlorophosphate was combined with ethanol (1 eq.) in dry CH2Cl2 containing 6a (1 eq.) at room temperature (20 oC), a half-life time for the formation of dimethylethylphosphate was determined as 100 hours (Scheme 4). Complex 6b gave an identical result, but when these imidazoles were replaced by NEt3 no reaction product was observed. The half-life time could be cut by carrying out the reaction at 40 oC, and under these conditions no reaction was observed when 6b was again replaced by NEt3. However attempts to use 10 mol % of either 6a/b as a true catalyst in the presence of NEt3 failed to give a measurable conversion to product within a meaningful time span. In comparison, use of one equivalent of 2-(9-anthryl)-1-(1-(tert-butyl)ethyl)imidazole 9 as promoter resulted in a half-life of 65 minutes, a time which further reduced to 27 minutes with the use of one equivalent of N-methylimidazole.

Scheme 4
Table 1. Reaction between ethanol and dimethylchlorophosphate
|
Conditions |
Half-life |
|
1 eq. 6a (20 oC) |
100 hours |
|
1 eq. 6b (20 oC) |
100 hours |
|
1 eq. NEt3 (20 oC) |
no reaction (after 120 hours) |
|
1 eq. 6b (40 oC) |
50 hours |
|
1 eq. NEt3 (40 oC) |
no reaction (after 65 hours) |
|
1 eq. 9 (20 oC) |
65 min. |
|
1 eq. NMI (20 oC) |
27 min |
New Ligands for Asymmetric Catalysis
We also sought to use these complexes for the rapid synthesis of ligands capable of acting as planar chiral mimetics. As oxazolines have been extensively used as ligands in asymmetric synthesis we decided to explore the potential of simple bidentate derivatives of 1. Thus again starting from carboxylic acid 4, amide 9 was readily obtained via reaction of the intermediate acid chloride with (S)-serine methyl ester hydrochloride in the presence of triethylamine. Dehydrative ring-closure was achieved with DAST7 to give oxazoline 10 in 93% overall yield (Scheme 5). From this three new ligands 11 - 13 were readily generated with each containing a b -hydroxyl group relative to the oxazoline nitrogen, an arrangement essentially identical to the b -hydroxy amines that have been extensively used in conjunction with dialkyl zinc reagents.8
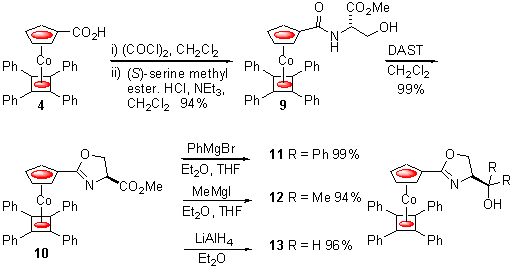
Scheme 5
Addition of 1.5 equivalents of diethyl zinc to benzaldehyde 14 in the presence of 5 mol% of these ligands resulted in the clean formation of 15, the highest selectivity of (R) to (S) enantiomers being obtained with the parent hydroxymethylene complex 13 (Table 2, entry 3). In contrast, phenyl and methyl substituents adjacent to the hydroxyl group in 11 and 12 result in erosion of selectivity correlated to the size of these alternatives to hydrogen (entries 1 and 2).

Scheme 6
Table 2. Addition of diethyl zinc to benzaldehyde.
|
Entrya |
Catalysta |
e.e. of 15.c (config.)d |
|
1 |
11 |
8 (R) |
|
2 |
12 |
54 (R) |
|
3 |
13 |
68 (R) |
|
4 |
14 |
75 (R) |
a) All reactions gave > 90% conversion after 24 h. b) 5 mol% with 1.5 eq. of Et2Zn. c) Determined by GC. d) Determined by GC comparison to commercial (R)-1-phenylpropanol.
Thus we reasoned that an increase in selectivity might alternatively relate to the size of the floor as defined by the metallocene phenyl substituents. To increase the number of these from four to five required a switch to the chemistry of pentaphenylferrocene 16.9 Functionalisation of this complex proved to be straight forward as reaction with 2-chlorobenzoyl chloride and aluminium chloride cleanly gave aryl ketone 17. This in turn was hydrolysed to give pentaphenylferrocene carboxylic acid 18 in 85% overall yield. Use of this as a starting material for the same chemistry as described above cleanly gave the novel oxazoline complex 19, again in excellent overall yield (Scheme 7). Application of this new ligand to the diethyl zinc/benzaldehyde reaction as before led to an increase in selectivity for the (R)-enantiomer (Table 2, entry 4).

Scheme 7
The absolute configuration of the 1-phenylpropanol resulting from these reactions may be rationalised by considering the two alternative reaction pathways A and B. Orientation of the oxazoline 4-substituent away from the floor defined by the phenyl groups results in a preference for the oxazoline rotamer drawn in both A and B. Following coordination to zinc, the ethyl group to be transferred may be oriented either away (A) or towards (B) the alkoxymethylene arm of the oxazoline. In the former, coordination of benzaldehyde from the side opposite to the floor results in ethyl transfer to the Re face and formation of the major R-enantiomer. In the alternative B the coordinated benzaldehyde is in close proximity to the bulky floor unless the oxazoline has rotated into a less stable conformation.
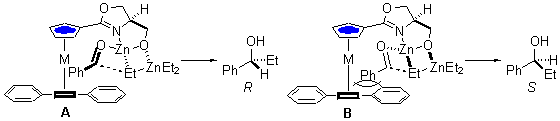
It is the ability of the phenyl groups to block one face of the heterocycle that enables the comparison with planar chirality to be made. The arrangement of groups is equivalent to the oxazoline itself being p -bonded to a metal/ligand fragment.
Conclusion
We have demonstrated that bulky metallocene appended imidazoles and oxazolines are readily synthesised, and that the heterocycles may be regarded as being in an environment of virtual planar chirality. Although the size of the metallocene has a significant effect on attenuating the activity of the imidazole complexes as nucleophilic catalysts, it is anticipated that the chiral relay principle demonstrated here will be readily extended to related complexes displaying significantly higher activity. Furthermore, the use of this approach for the rapid synthesis of new ligands has been demonstrated, and the use of related systems in reactions of wider applicability is currently under investigation.
Acknowledgements
We wish to thank the EPSRC for financial support (GR/M55893), Abdul Malik and Jamie Platt for the X-ray structure determination, Dave Butler for the synthesis of 18, and finally Rob Jenkins for his invaluable day-to-day assistance.
References and Notes
All comments on this poster should be sent by e-mail to (mailto:[email protected])
[email protected] with a0092 as the message subject of your e-mail.