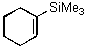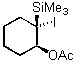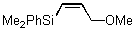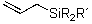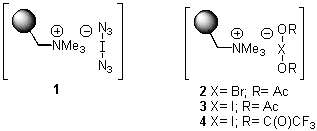
Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[B0001]
Cohalogenation of Allyl- and Vinylsilanes usingSilvie Domann, Georgia
Sourkouni-Argirusi, Nuria Merayo, Andreas Schonberger
and Andreas Kirschning*
Institut fur Organische Chemie der
Technischen Universitat Clausthal, Leibnizstrasse 6, D-38678 Clausthal-Zellerfeld,
Germany
Tel.: int. (+)-(0)5323-72 3886; Fax: int. (+)-(0)5323-72 2858; E-mail: [email protected]
Received: 2 August 2000 / Uploaded: 3 August 2000
_________________________
Abstract: Polymer-supported electrophilic halogenate(I) complexes 2 and 3 promote smooth addition to vinyl and allylsilanes without loss of the silyl group. In conjunction with Amberlyst A26 (OHs -form) vinyl silanes are converted into epoxysilanes in one pot.
__________
I. Introduction
Recently, the development and applications of polymer-supported reagents have seen a dramatic increase in interest.1 Functionalized matrices can be used in excess to drive reactions in solution to completion and are finding application in high throughput, automated parallel syntheses.2 In some cases major differences between reactions on polymer supports and their low-molecular mass analogues have been observed.3 Apart from differences in reaction rates, altered regio- and stereoselectivity has been described in some cases using functionalized polymers. E. g. Patchornik and coworkers encountered that crosslinked poly(maleimide) in which 70% of the repeat units were N-brominated gave a different set of bromination products in reactions with cumene than N-bromosuccinimide.4 These properties give polymer-supported reagents additional attraction. Thus, in this communication we report on the unexpected clean cohalogenation of allyl and vinylsilanes which is promoted by functionalized polmers with electrophilic properties.
Recently, our interest in new electrophilic halogen-ate(I) complexes5 has led to the development of the first polymer-bound iodine azide source 16 as well as of functionalized polymers 2 - 4 wich are loaded with synthetic equivalents of acylated hypohalites.7 All of these electrophilic reagents promote 1,2-cohalogenations of various alkenes8,9 under very mild conditions and with high efficiency.7
II. Results and Discussion
In this paper, we disclose reactions of vinyl and allylsilanes with polymer-supported electrophilic reagents 2 and 3 which instead to the expected desilylation lead to 1,2-addition products. In fact, numerous methods10 are known for effecting iododesilylation of vinyl and allylsilanes using different iodonium sources like ICl, IBr, IBF4, and NIS.11 It is generally accepted that the reaction is initiated by attack of the electrophilic iodonium source to the double bond to form a cyclic iodonium ion and formation of b-silicon stabilized cation. After rotation around the C-C-bond a planar orientation between the empty p-orbital and the C-Si bond is achieved which allows facile removal of the silyl group by a nucleophile. In some instances, additional stabilization of the intermediate carbocation by the solvent was accounted.11
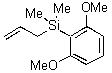
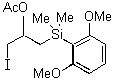
In contrast to these results, we observed exclusive formation of the 1,2-addition products 11 - 13 when vinylsilanes 5 and 6 were treated with polymer-supported reagents 1 - 3 in dichloromethane (Table 1). We observed that dimethylphenylsilyl-substituted alkene 6 reacted much slower than 1-trimethylsilyl-cyclohexene 5. In a similar manner the even more reactive allylsilanes 7 - 9 gave 1,2-cohalogenation products 14 - 16 in good yield.12 Only alkynylsilane 10 was unreactive under the conditions typically employed.13 This latter result is in sharp contrast to the treatment of terminal alkynes with functionalized polymer 3 which affords alkynyl iodides.7b
As an extensions of these studies, we utilized iodate(I) reagent 3 and Amberlyst A26 (OHs - form) for developing a simple one pot addition and cyclization protocol. Starting from vinylsilanes 5 and 6, crude 1,2-cohalogenation products 11 and 12 were further transformed under basic conditions to the corresponding epoxysilanes 17 and 18 in good yield and with high purity (Scheme 1).
Table 1. Haloacetoxylation of vinyl and allyl silanes.
Alkene |
Reagenta (equiv.)b |
Product |
Yield %,c Purity % |
||||
|
|
|
|||||
5 |
3 (4), 5h |
|
11 | 76 (80), >99 |
|||
|
|
|
|||||
6 |
3 (6), 60h 2 (4), 40h |
X= I X= Br |
12 13 |
84 (98), >99 77 (90)d |
|||
|
|
 |
|
||||
7 8 |
R= Me, R?= Ph R= Ph, R?= Me |
3 (4), 6h 3 (3), 6h |
Nu= OAc Nu= OAc |
14 15 |
62 (75), >99 67 (80), >99 |
||
|
|
|
|
|||||
9 |
3 (5), 2.5h |
16 | 83, >95 |
||||
 |
 |
-------- |
|||||
10 |
3 (8), 8d |
no reaction |
|||||
a
For experimental details refer to reference 13. b All reagents are employed in excess with reference to the amount of polymer-bound halide specified by the commercial provider.14 c All yields refer to isolated pure products. Yields in brackets refer to crude products. d Formation of the 1,2-cis-addition byproduct was proven by 1H NMR-spectroscopy of the crude product.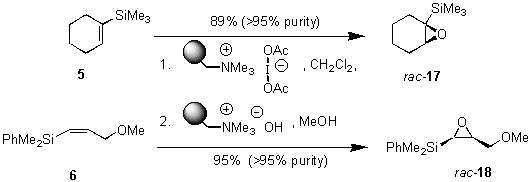 Scheme 1
Scheme 1
In conclusion, polymer-supported haloate(I)-complexes promote cohalogenation
of allyl and vinylsilanes under very mild conditions without exerting desilylation. These reagents allow easy product isolation, are conveniently recycled15 and finally are potentially useful for automated solution phase synthesis.Acknowledgement: Support by the Fonds der Chemischen Industrie and by Novabiochem (Switzerland) is gratefully acknowledged. We thank Prof. Dr. E. Schaumann for providing us with vinyl and allylsilanes.
III. References and Notes
(1) Reviews: (a) Shuttleworth, S. J.; Allin, S. M., Sharma, P. K., Synthesis 1998, 1217-1239. (b) Kaldor, S. W.; Siegel, M., G., Curr. Opin. Chem. Biol., 1997, 1, 101-106. (c) Hodge, P.; Sherringtom, D. C. In Polymer-Supported Reaction in Synthesis, Wiley: New York, 1990. (d) Laszlo, P., Ed., In Preparative Chemistry using Supported Reagents, Academic, San Diego, 1987.
(2) (a) Flynn, D. L.; Devraj, J. J. Parlow, Curr. Opin. Drug Discovery Dev. 1998, 1, 41-50. (b) Suto, M. J.; Gayo-Fung, L. M.; Palanki, M. S. S.; Sullivan, R., Tetrahedron 1998, 54, 4141-4150. (c) Parlow, S. S., Tetrahedron Lett. 1996, 37, 5257-5260. (d) Habermann, J.; Ley, S. V.; Scott, J. S., J. Chem. Soc., Perkin Trans I, 1998, 3127-3130. (e) Caldarelli, M.; Habermann, J.; Ley, S. V., J. Chem. Soc., Perkin Trans. I, 1999, 107-110.
(3) Hodge, P., Chem. Soc. Rev. 1997, 26, 417-424.
(4) Yaroslavsky, C.; Patchornik, A.; Katchalski, E., Tetrahedron Lett. 1970, 3629-3632.
(5) (a) Kirschning, A.; Plumeier, C.; Rose, L., Chem. Commun., 1998, 33-34. (b) Hashem, Md. A.; Jung, A.; Ries, M.; Kirschning, A., Synlett 1998, 195-197. (c) Kirschning, A.; Hashem, Md. A.; Monenschein, H.; Rose, L.; Schoning, K.-U., J. Org. Chem., 1999, 64, 2720-2722.
(6) Kirschning, A; Monenschein, H.; Schmeck, C., Angew. Chem. 1999, 111, 2720-2722; Angew. Chem. Int. Ed. Engl, 1999, 38, 2594-2596.
(7) (a) Kirschning, A.; Jesberger, M.; Monenschein. H., Tetrahedron Lett. 1999, 40, 8999-9002. (b) Monenschein, H.; Sourkouni-Argirusi, G.; Schubothe, K. M.; O?Hare, T.; Kirschning, A., Org. Lett. 1999, 1, 2101-2104.
(8) For a recent example for iodate(I)-promoted iodination of arenes refer to S. Tripathy, R. LeBlanc, T. Durst, Org. Lett. 1999, 1, 1973-1975.
(9) Recently, an interesting new method for 1,2-coiodination of alkenes using dimethyldioxirane-promoted oxidation of iodomethane was disclosed by G. Asensio, C. Andrea, C. Boix-Bernardini, R. Mello, M. E. Gonzalez-Nunez, Org. Lett. 1999, 1, 2125-2128.
(10) (a) Chan, T. H.; P. W. Lau, W. Mychajlowskij, Tetrahedron Lett. 1977, 18, 3317-3320. (b) Miller, R. B.; Reichenbach, J., Tetrahedron Lett. 1974, 15, 543-546. (c) Miller, R. B.; McGarvey, G.; J. Org. Chem. 1978, 43, 4424-4431. (d) Chan, T. H.; Fleming, I., Synthesis 1979, 761-786. (e) Huynh, C.; Linstrumentelle, G., Tetrahedron Lett. 1979, 20, 1073-1076. (f) Tamao, K.; Akita, M.; Maeda, K. Kumuda, M., J. Org. Chem. 1995, 36, 2153-2156. (g) Barluenga, J.; Alvarez-Garcia, L. J.; Gonzalez, J. M., Tetrahedron Lett. 1995, 36, 2153-2156.
(11) Stanos, D. P.; Taylor, A. G.; Kishi, Y., Tetrahedron Lett. 1996, 37, 8647-8650 and references cited therein.
(12) We collected evidence that the binary compound [I-OAc] is the active electrophilic reagent, which is slowly released from the resin 3: Monenschein, H.; Kirschning, A. unpublished results.
(13) The preparation of polymer-bound reagents 3 is described in references 6 and 7.
General procedure for the 1,2-cohalogenation of silylated alkenes: A mixture of alkene (1 equiv.) and resin (for number of equiv. refer to Table 1) were shaken at 300 rpm under light protection in dry CH2Cl2 (1.5 ml / mmol) at rt. Completion of the reaction was monitored by tlc. Filtration terminated the reaction. The resin was washed with CH2Cl2 (3x) and the combined organic washings and filtrate were concentrated under reduced pressure. In some cases, further purification by column chromatography was necessary.
(14) This fact, may either be rationalized by assuming that only a proportional amount of immobilized halide was transformed into the haloate(I)-species or that only the most accessible haloate(I) anions are involved in the cohalogenation process. If acylated hypohalites are the active species after release from the polymer (see also reference 12), their degradation prior to the reaction with alkenes may also contribute to the need for a formal excess of reagent.
(15) The functionlized polymers were recycled to the halide form without loss of activity by treatment with concentrated aqueous HBr or HI for 1 h at rt.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with B0001 as the message subject of your e-mail.