[B0013]
Solid-Phase Synthesis of Peptides, Peptidomimetics, and Cyclic Peptides Using Traceless Aromatic Side-Chain Tethered Building Blocks
Younghee Lee and Richard B. Silverman*
Department of Chemistry
Northwestern University
Evanston, Illinois, USA 60208-3113
Received: 31 August 2000 / Uploaded: 31 August 2000
Introduction
Combinatorial chemistry, combined with recent advances in robotic screening, which enable the testing of a large number of compounds in a short period of time, is becoming an important tool in accelerating drug discovery. This technique involves the preparation of a large number of structurally related compounds either as mixtures in the same reaction vessel or individually by parallel synthesis. In this manner large pools of similar compounds can be synthesized within a short period of time. Combinatorial libraries have been prepared using both solution chemistry and by solid phase synthesis; however, solid phase synthesis allows the use of excess reagents to drive the reaction to completion and easy removal of the reagents and side-products by simple filtration of the polymeric support and washing with solvent. Therefore, solid phase synthesis offers a more attractive approach to the generation of chemical libraries for screening purposes.
One of the key elements in solid phase chemistry is the polymeric resin. Since the introduction of the Merrifield resin, libraries of peptides, nucleotides, and organic molecules have been generated on solid supports. Many of the resins that are currently employed as supports were originally developed for the synthesis of peptides. Polar functionalities such as carboxylic acids and amides were released upon cleavage of products from the resins. Recent advances in linker technology have allowed other polar functional groups, such as alcohols and thiols, also to be attached to the polymer support. In fact, most of the linkers available for solid support synthesis to date require polar functional groups for binding, and the same polar groups are released after cleavage. To generate libraries with biological activity, however, such polar functionalities may possess unfavorable pharmacological properties. Because of poor oral bioavailability and enzymatic degradation of linear peptides, modified peptides, peptidomimetics, and cyclic peptides have become appealing targets for the design of therapeutic agents with increased pharmacological activities. Therefore, we directed our attention to the design of new solid support linkers for non-polar or aromatic compounds that are commonly found in medicinally important agents.
Conventional solid-phase peptide synthesis allows the elongation of the amino acid backbone unidirectionally (C to N or N to C). However, attachment of the amino acid side chain to the polymer would permit chain elongation in both directions; thus, more diverse libraries of peptides and peptidomimetics could be prepared than with conventional methods. This approach appears to be a method of choice for compounds whose C- and N-terminals are both capped with non-amino acids. Unfortunately, side chain anchoring of amino acids to the polymer support for solid phase synthesis has been applicable only to amino acids which have polar side chains such as Asp and Glu (COOH), Cys (SH), Ser and Tyr (OH), Lys (NH2), or His (imidazole). This is because of the limitation of current linkages for solid supports. Most of the linkages used for linear or cyclic peptide synthesis on solid support are based on polar functional groups. For the amino acids that have a hydrophobic aliphatic or aromatic carbon chain, a side chain anchoring strategy for solid phase synthesis had not been reported until our recent work, some of which is discussed in this electronic lecture.
Because of the nonpolar nature and steric bulkiness of its side chain, phenylalanine is one of the preferred residues in peptidomimetics when the biological targets are known to have hydrophobic binding sites. For example, almost every aspartyl protease for which substrate specificities have been studied (e.g., HIV protease, renin, cathepsins D and E, etc.) has a preference for hydrophobic amino acid side chains at the P1 position. It is not surprising that most of the HIV protease inhibitors on the market or in clinical studies have phenylalanine or other bulky hydrophobic groups at the P1 position. Also, to increase the oral bioavailability of compounds derived from peptidomimetic approaches, amino acid residues with bulky and hydrophobic side chains are often left unchanged where other residues are modified. In this regard, phenylalanine or other amino acids with nonpolar aromatic side chains are considered to be key pharmacophores in many biologically important peptide-like molecules.
To design a library of oligopeptides and peptidomimetics containing phenylalanine as the key pharmacophore, the most efficient way would be to attach the phenylalanine side chain to the polymer support and to vary the residues at both the N- and C-termini. However, to the best of our knowledge, there has been no previous research for anchoring aromatic side chains of phenylalanine to a polymer support.Several novel strategies utilizing a resin-bound arylsilane as a traceless linker have been developed for the solid phase synthesis of aromatic or heteroaromatic compounds. This technology allows the attachment of substrates to the support at an inert site within the molecule. Upon cleavage from the resin by protiodesilylation, no trace or memory of attachment to the polymer support remains on the released product. Also, silicon-directed ipso substitution of arylsilanes is frequently used for regiospecific introduction of electrophilic functional groups such as bromine and iodine to the aromatic ring.
Libraries of compounds constructed on solid support using silyl linkages include 1,4-benzodiazephines,6a biaryls,6c benzofurans,6c and tricyclics.6d We reasoned that an appropriate arylsilyl linkage could be used for tethering phenylalanine precursors to the polymer. The combination of orthogonal protection schemes using an Fmoc/Ca-allyl amino ester would permit the preparation of more complex peptides because allyl esters can be hydrolyzed specifically with Pdo[P(Ph3)]4 under nearly neutral conditions without affecting other protecting groups. After building both the N- and C-termini of amino acids, protiodesilylation or ipso-substitution of the silyl group would cleave the desired phenylalanine-containing product from the resin. This approach is discussed in this electronic lecture for the synthesis of phenylalanine- and 3-aryl b-amino acid-containing peptides.Head-to-tail cyclization of peptides on the resin provides a facile route to cyclic compounds. In addition to general advantages of solid phase synthesis, such as high efficiency and easy purification, head-to-tail cyclization of peptides on polymer supports provides minimal risk of intermolecular reactions (e.g., dimerization and oligomerization), even under high concentration. This is another advantage over solution chemistry which requires high dilution conditions to avoid intermolecular side reactions of the linear peptide. The synthesis of cyclic peptides via a traceless aromatic side-chain attachment with head-to-tail cyclization on the solid support also will be discussed.
Phenylalanine-containing peptides
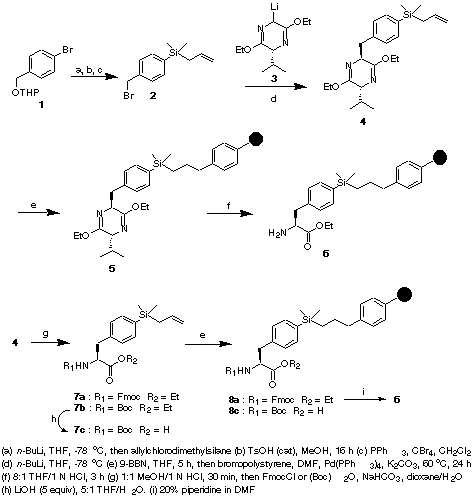
An appropriate arylsilyl linkage could be used for tethering phenylalanine to the polymer via its phenyl group, then further elongation of the amino acid backbone would allow rapid synthesis of phenylalanine-containing molecules. Scheme 1 illustrates the synthesis of a phenylalanine derivative having a silyl linker attached to the phenyl ring, and the loading of this compound to brominated polystyrene resin. Lithium-halogen exchange of THP-protected 4-bromobenzyl alcohol 1 with n-BuLi in THF followed by treatment with allylchlorodimethylsilane afforded the corresponding 4-allyldimethylsilyl substituted compound. Deprotection of the THP group with catalytic p-toluenesulfonic acid in MeOH overnight, and subsequent reaction of the resulting alcohol with PPh3/CBr4 in CH2Cl2 generated 2. A carbon-carbon bond forming reaction
Scheme 1
between 2 and the lithitiated Schollkopf bislactim ether 3 in THF at -78
oC provided a mixture of diastereomers in a ratio of 94:6. The major diastereomer (4) was isolated in 71% yield after silica gel column chromatography. The Schollkopf adduct 4 was attached to the polymer in a two-step sequence. Hydroboration of the allylsilane (9-BBN in THF), followed by in situ Suzuki coupling of the borane complex with bromopolystyrene resin [Pd(0), K2CO3, in THF], provided the resin bound phenylalanine precursor 5. Treatment of 5 with a solution of THF/1N HCl (8:1) for 3 h at room temperature afforded polymer-bound phenylalanine ethyl ester 6, which is ready for further derivatization. Alternatively, 4 can be hydrolyzed under mild acidic conditions (1:1 MeOH/1 N HCl), and the resulting amine protected either as an N-Fmoc carbamate (FmocCl, NaHCO3) (7a) or an N-Boc carbamate ((Boc)2O, NaHCO3) (7b); saponification of 7b with LiOH in THF/H2O gives 7c. Hydroboration and Suzuki coupling of carbamates 7a and 7c with bromopolystyrene resin under the conditions described above provided the resin bound phenylalanine derivatives 8a and 8c, respectively. The loading level of the amino acid derivatives bound to the polymer can be determined by the Fmoc release UV/vis assay or by mass balance of the corresponding products released from the resin after treatment of the resin with TFA or Br2 in dichloromethane.The utility of the polymer-bound phenylalanine precursors was demonstrated by the preparation of dipeptide analogues, from both the N- and C-termini. As shown in Scheme 2,
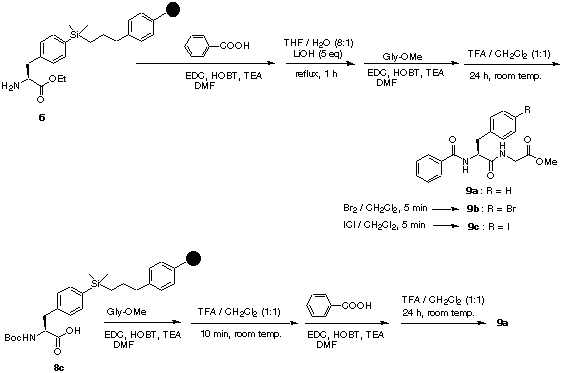
Scheme 2
coupling (EDC, HOBT, TEA in DMF) of 6 with benzoic acid formed the N-terminal amide. Hydrolysis of the ester did not proceed at all under the standard conditions (LiOH [5 equiv] in THF/H2O (8:1), 16 h at room temperature), possibly because of poor swelling properties of the bromopolystyrene resin under the reaction conditions. However, the desired hydrolysis proceeded to completion when the same solution was heated to reflux for 1 h. C-Terminal amide coupling was performed on this carboxylic acid with glycine methyl ester under the conditions described above. After completion of the synthesis, cleavage of the desired dipeptide analogue from the resin was carried out with 50% TFA in dichloromethane for 24 h, giving protected dipeptide 9a in 93% yield. The purity of the crude sample was determined to be about 95% by 1H NMR spectroscopy. Alternatively, cleavage of the dipeptides from the resin by ipso-substitution of the silyl group with electrophiles (Br2 or ICl in dichloromethane for 5 min) provides the corresponding halogen-substituted compounds (9b, 97%; 9c, 94%) with purities higher than 95% as determined by 1H NMR.
Similarly, but in a reversed reaction sequence, the same compound (9a) was synthesized on the solid support starting from the N-Boc-protected phenylalanine precursor 8c. C-Terminal amide coupling with glycine methyl ester followed by deprotection of the Boc group with 50% TFA in dichloromethane for 10 min, and subsequent coupling of the resulting amine with benzoic acid afforded 9a in 91% yield after cleavage from the resin (50% TFA in CH2Cl2 for 24 h). These two operations demonstrate that small phenylalanine containing peptides can be prepared on a solid support in any order of reaction sequence desired. The polymer-bound phenylalanine can be modified in either the N- or C-terminal direction with readily available reagents, such as amines and carboxylic acids, and diversity can be further increased by cleavage of the peptide from the resin with halogens to give p-substituted halophenylalanines. These halogenated analogues, then, can be even further elaborated by a variety of alkyl- or aryl9
substitution reactions at the halide.The arylsilyl linkage was found to be resistant to moderate acidic [1 N HCl/THF (1:8); 50% TFA/ CH2Cl2 for 10 min] and basic conditions [LiOH, THF/H2O (8:1), heat] as well as to the general amide coupling reactions. Both N-Boc and N-Fmoc-protection strategies employed for the preparation of dipeptides 9 indicate that this versatility may be of great value for the solid-phase synthesis of complex molecules requiring various orthogonal protection of amine intermediates.
Phenylglycine-containing peptides
Not only phenylalanine derivatives but also other amino acids with aromatic side chains are frequently seen in both natural and synthetic peptides of interest. For example, phenylglycine is an important component of many natural products. One of the phenylglycine containing cyclic peptides of biological significance is the family of pristinamycin I. Pristinamycin I, along with its other synergistic component pristinamycin II, is used for the treatment of infection.
The therapeutic applications of this antibiotic are likely to increase because of the increased incidence of resistance to other commonly used antibiotics. The same arylsilyl linkage discussed earlier for phenylalanine derivatives could be employed for tethering phenylglycine.To prepare a phenylglycine derivative with a silyl group attached to the phenyl ring, an Evans asymmetric azidation can be used to introduce an azide at the
a-carbon of the acid. Because the reduction of the azide to the amine is well established both in solution and by solid phase synthesis, the azido group is thought to be a suitable amine equivalent. As shown in Scheme 3, the key intermediate for the azido analogue of the phenylglycine derivative, 4-(allyldimethylsilyl)phenylacetic acid (13) was prepared from 1,4-dibromobenzene (10). A metal-halogen exchange reaction of the starting 1,4-dibromobenzene with n-butyllithium followed by treatment with allyldimethylsilyl chloride installed the silyl function onto the benzene ring (11). Lithiation with t-butyllithium followed by reaction with ethylene oxide provided the two-carbon homologated compound 12. Oxidation with Jones reagent converted the alcohol 12 to the carboxylic acid 13. This acid was activated as a pivaloyl anhydride which was treated with lithiated oxazolidinone 14 to obtain the imidate intermediate 15. Asymmetric azidation was carried out by sequential treatment of imidate 15 with KHMDS followed by trisyl azide, resulting in azidoimidiate 16 as a single diastereomer after purification. Titanium(IV)-mediated esterification in methanol gave ester 17, which was converted to the Boc/ester protected phenylglycine derivative (18). Suzuki coupling of the allyl group to bromopolystyrene produced the side-chain tethered phenylglycine derivative (19). This building block can be elaborated in either the N- or C-direction to make phenylglycine-containing compounds. For example, following Boc-deprotection with TFA, benzoic acid was coupled onto the amino group and the product (20) was released from the resin by ipso-bromo substitution (Scheme 4).
Scheme 3

Scheme 4
3-Aryl-b-amino acid-containing peptides
If libraries of molecules require an aromatic side chain as a pharmacophore, an a-amino acid can be replaced with other types of multi-functional units, such as b-amino acids. New amino acid building blocks can be designed to generate libraries of compounds containing the aromatic side chain. As an example, a cyclo-b-tetrapeptide prepared by Seebach�s group as a somatostatin analogue was found to display significant biological activity and affinity for human receptors. This suggests that an amino acid backbone can be replaced by other structures, supporting peptidomimetic approaches for peptide analogues.
b-Amino acids also are frequently found in natural products and in therapeutic agents. Because of the enzymatic stability of b-peptides in biological systems, b-amino acids are useful building blocks for the design of new peptidomimetics. Although there are several known synthetic methods for b-amino acids, such as the Arndt-Eistert homologation of a-amino acids and catalytic hydrogenation of 3-aminoacrylate, and a number of b-amino acids are commercially available, it is highly desirable to develop polymer-bound b-amino acid building blocks for the rapid synthesis and high diversification of compounds.
Recently, a practical and highly enantioselective synthesis of tert-butanesulfinamide (21) was reported by the Ellman group. This chiral building block was used effectively for the asymmetric synthesis of b-amino acids. It was also found that the tert-butanesulfinyl group acts as a Boc surrogate, which is advantageous for solid-phase reactions. This method for b-amino acid synthesis using tert-butanesulfinamide is generally applicable, and we found that it was well adapted for the asymmetric synthesis of b-amino acid analogues with an aromatic side chain substituted with a silyl group, which was attached to a polymer support. As shown in Scheme 5, commercially-available 4-bromobenzaldehyde (22) was treated with ethylene glycol with a catalytic amount of p-toluenesulfonic acid to afford the 1,3-dioxolane derivative of 22. Lithium-halogen exchange of the intermediate with t-butyllithium at �78 ˚C followed by addition of allyldimethylsilyl chloride afforded 23 in an 86% yield from 22. Refluxing of 23 in acetone in the presence of p-toluenesulfonic acid (cat) for 6 h provided 4-allyldimethylsilylbenzaldehyde (24) in an 86% yield. Alternatively, 24 was synthesized from 1,4-dibromobenzene (25) in a 67% yield by a two step sequence: (1) replacement of one bromine with a silyl group and (2) replacement of the other bromine using a formylation reaction (t-BuLi, THF, -78 ˚C, then DMF). Condensation of 24 with (R)-(-)-tert-butanesulfinamide was performed in the presence of titanium propoxide in refluxing THF for 1 h to give the tert-butanesulfinyl imine 26 as an oil in a 68% yield. The titanium enolate generated by transmetalation of lithiated methyl acetate with ClTi(O-i-Pr)3 in THF at -78 ˚C was allowed to react with 26 for 3 h to provide 27 in a 79% yield. The diastereoselectivity of 27 was determined by the analysis of the Mosher amide (28), prepared by deprotection of the tert-butanesulfinyl group (4 N HCl/dioxane, MeOH) followed by subsequent derivatization of the amino group with (R)-(-)-a-methoxy-a-(trifluoromethyl)-phenylacetic acid chloride (MTPACl). Analysis of both 1H NMR and19F NMR spectra of 28 showed less than 1% of the minor diastereomer, which suggests that titanium enolate addition to the sulfinyl imine 26 proceeded in high stereoselectivity. Hydroboration of the terminal olefin of 27 with 9-BBN in THF followed by in situ Suzuki coupling9 of the borane complex with bromopolystryrene resin10 [Pd(PPh3)4, Na2CO3, in THF/DMF] resulted in polymer bound b-amino acid derivative 29. The loading level (0.32 mequiv/g) of 29 was determined by mass balance of (3R)-methyl 3-amino-3-(4-bromophenyl)-butyrate (30), which was obtained by stirring an aliquot of resin 29 with 50% TFA in CH2Cl2 for 5 min followed by washing and then the cleavage reaction (Br2, CH2Cl2, 20 min).

Scheme 5
Conditions: (a) ethylene glycol, p-toluenesulfonic acid (cat), benzene, reflux, 4 h; (b) t-BuLi, THF, -78
oC, then allylchlorodimethylsilane; (c) acetone, p-toluenesulfonic acid (cat), reflux, 6 h (d) n-BuLi, THF, -78 oC, then allylchlorodimethylsilane; (e) t-BuLi, THF, -78 oC, then DMF; (f) Ti(OPr)4, THF, reflux, 1 h; (g) methyl acetate, LDA, THF, -78 oC, then chlorotitanium triisopropoxide (2.1 eq); (h) 4 N HCl/dioxane, MeOH, 5 min; (i) (R)-MTPACl, pyridine, CHCl3, 2 h; (j) 9-BBN, THF, 5 h, then bromopolystyrene, DMF, Pd(PPh3)4, Na2CO3, 75 oC, 48 h. (k) 50% TFA in CH2Cl2, 5 min; (l) Br2 in CH2Cl2, 20 min.To demonstrate the suitability of building block 29 in solid-phase synthesis,
b-amino acid-containing tripeptides 32 were synthesized according to the procedure shown in Scheme 6. Resin 29 was treated with 50% TFA in CH2Cl2 for 5 min to deprotect the amino group, which was treated with Fmoc-Ala-OH using standard EDC and HOBt coupling conditions in DMF, then the Fmoc group was removed with 20% piperidine in DMF for 30 min. After several washings of the resin, benzoic acid was coupled (EDC, HOBt, TEA in DMF) to the amine to construct polymer-bound dipeptide analogue 31. Hydrolysis of the ester group at the C-terminus of 31 was carried out with LiOH (5 eq.) in THF/H2O (8:1) under refluxing conditions for 2.5 h, and the resulting carboxylic acid was coupled with Gly-OEt under the same conditions described above (EDC, HOBt, TEA in DMF). Upon completion of the synthesis, cleavage of the product with 50% TFA in CH2Cl2 at room temperature for 24 h yielded 32a in an 88% yield. Alternatively, cleavage of the tripeptide from the resin by ipso-substitution of the silyl group with either Br2 or ICl in CH2Cl2 for 20 min afforded 32b and 32c, respectively, in 95% yields. In all cases, the purity of 12, as determined from the 1H NMR spectra of the crude products, was greater than 95%.
Scheme 6
Utilization of the aromatic side-chain tethered methodology for the synthesis of the natural cyclic depsipeptide sansalvamide
Sansalvamide (33) is a cyclic depsipeptide produced by a marine fungus of the Genus Fusarium found in Little San Salvador Island, Bahamas. Sansalvamide is composed of four

amino acids (Phe, 2 Leu, Val) and one hydroxy acid (O-Leu), with five chiral carbons all having S-stereochemistry, which was determined by an extensive analysis of NMR data and chiral capillary GC of the ester derivative of fragmented amino acids after acidic hydrolysis. This highly lipophilic natural product was found to have significant cancer cell cytotoxicity with a mean IC50 value of 27.4
mg/mL against the United States National Cancer Institute�s 60 cell-line panel.The synthetic method described earlier in this electronic lecture for the polymer-bound phenylalanine building block involved a C-C bond forming reaction (Scheme 1) of lithiated Schollkopf�s bislactim ether (3) with 4-allyldimethylsilylbenzyl bromide (2) as a key step which resulted in two diastereomers (4) in a ratio of 94 to 6. The tedious column chromatography procedure required for the separation of the major isomer led us to investigate an alternative synthetic route for the silylated phenylalanine building block as shown in Scheme 7. The

Scheme 7
Conditions: (a) methyl-2-(tert-butoxycarbonylamino)-2-(dimethoxyphosphinyl)-acetate, tetramethylguanidine, THF, -78
oC to rt, 86%; (b) [((S,S)-Et-DuPHOS)-Rh]+ (1 mol%), H2 (1 atm), CH2Cl2, 23 h, 100%; (c) (Boc)2O, DMAP (cat), THF, reflux, 1 h; (d) hydrazine, MeOH, 4 h, 93% for two steps; (e) 9-BBN, THF, rt, 5 h, then 37, DMF, Pd(PPh3)4, 2 N Na2CO3, 75 oC, 3 days; (f) Br2 in CH2Cl2, 20 min.; (g) 50% TFA in CH2Cl2, 15 min; (h) (R)-MTPACl (3 eq), DIPEA (4 eq), CH2Cl2, 4 h.Horner-Emmons reaction with methyl-2-(tert-butoxycarbonylamino)-2-(dimethoxyphosphinyl)-acetate, tetramethylguanidine gave the (Z)-enamide ester exclusively. The key step is the utilization of commercially-available [((S,S)-Et-DuPHOS)-Rh]
+ catalyst system in CH2Cl2 under H2 (1 atm) to create the desired chiral a-carbon in quantitive yield without affecting the terminal olefin. Conversion of the N-acetyl group to N-Boc was carried out in one pot by treatment of the chiral intermediate with Boc2O in the presence of a catalytic amount of DMAP in refluxing THF. The loading level of 38 was determined to be 0.12 mmol/g by measuring the mass balance after bromine cleavage from the resin. Analysis of both 1H and 19F NMR spectra of the Moser amide indicated a high enantiopurity (>98% ee) of polymer-bound building block 38.Scheme 8 shows the solid-phase synthesis of sansalvamide (33) from 38. After ester hydrolysis, the resulting acid was treated with H-Leu-OLeu-Oallyl (44, synthesized in three steps from leucine) under the standard amide coupling conditions (HBTU, DIPEA, NMP) to give 40. Following deprotection of the N-Boc protecting group, extension of the N-terminus using Fmoc

Scheme 8
Conditions: (a) LiOH (5 eq), THF/H2O (7:1), rt, 16 h; (b) HBTU (4 eq), DIPEA (4 eq), 44 (4 eq), NMP, 16 h; (c) 50% TFA in CH2Cl2, 15 min; (d) Fmoc-Leu-OH (4 eq), HBTU (4 eq), DIPEA (4 eq), NMP, 6 h; (e) 20% piperidine in DMF, 40 min; (f) Fmoc-Val-OH (4 eq), HBTU (4 eq), DIPEA (4 eq), NMP, 6 h; (g) CHCl3/AcOH/NMM (37:2:1), Pd(PPh3)4 (4 eq), 3 h; (h) HBTU (4 eq), DIPEA (4 eq), NMP, 16 h; (i) 50% TFA in CH2Cl2, 36 h; (j) allyl bromide, K2CO3, acetone, 48 h, 95%; (k) Boc-Leu-OH, DMAP (cat), DCC, CH2Cl2, 63%; (l) 50% TFA in CH2Cl2, 5 min.
strategy in three consecutive reaction sequences, a protected linear intermediate 41 was produced. Deprotection of the ester and amine functionalities gave a polymer-bound depsipeptide, which was cyclized with HBTU and DIPEA in NMP. Sansalvamide (33) was obtained by TFA cleavage from the solid support in a 67% overall yield for the 12 solid-phase steps, based on the initial loading level of 38. A comparison of the 1H NMR spectra of synthetic sansalvamide and a sample of the natural product (donated by Professor William Fenical, Scripps Institution of Oceanography) indicated that the synthetic material was identical to that of the natural product with >95% purity.
These results indicate that the polymer-bound phenylalanine building block methodology can provide rapid access to cyclic depsipeptides (and cyclic peptides) consisting of hydrophobic side chains. Furthermore, the arylsilyl linkage is compatible with a variety of reaction conditions and reagents, but can be cleaved by several different conditions.
References
[1] (a) Hintermann, T.; Seebach, D. Chimia 1997, 50, 244. (b) Seebach, D.; Abele, S.; Schreiber, J.; Martinoni, B.; Nussbaum, A. K.; Schild, H.; Schulz, H.; Hennecke, H.; Woessner, R.; Bitsch, F. Chimia 1998, 52, 734.
[2] (a) Trzeciak, A.; Bannwarth, W. Tetrahedron Lett. 1992, 33, 4557-4560. (b) Sabatino, G.; Chelli, M.; Mazzucco, S.; Ginanneschi, M.; Papini, A. M. Tetrahedron Lett. 1999, 40, 809-812. (c) Rovero, P.; Quartara, L.; Fabbri, G. Tetrahedron Lett. 1991, 32, 2639-2642. (d) Kates, S. A.; Sole, N. A.; Johnson, C. R.; Hudson, D.; Barany, G.; Albericio, F. Tetrahedron Lett. 1993, 34, 1549-1552. (e) Zhong, H. M.; Greco, M.N.; Maryanoff, B. E. J. Org. Chem. 1997, 62, 9326-9330. (f) Sugiura, T.; Hamada, Y.; Shioiri, T. Tetrahedron Lett. 1987, 28, 2251-2254.
[3] (a) Lee, Y.; Silverman, R. B. J. Am. Chem. Soc. 1999, 121, 8407-8408. (b) Lee, Y.; Silverman, R. B. Organic Lett. 2000, 2, 303-306.
[4] (a) Garrett, G. S.; McPhail, S. J.; Tornheim, K.; Correa, P. E.; McIver, J. M. Bioorg. Med. Chem. Lett. 1999, 9, 301-306. (b) Fink, C. A.; Carlson, J. E.; Boehm, C.; McTaggart, P.; Qiao, Y.; Doughty, J.; Ganu, V.; Melton, R.; Goldberg, R. Bioorg. Med. Chem. Lett. 1999, 9, 195-200. (c) Roush, W. R.; Gwaltney II, S. L.; Cheng, J.; Scheidt, K. A.; McKerrow, J. H.; Hansell, E. J. Am. Chem. Soc. 1998, 120, 10994-10995. (d) Roush, W. R.; Gonzalez, F. V.; McKerrow, J. H.; Hansell, E. Bioorg. Med. Chem. Lett. 1998, 8, 2809-2812. (e) Adams, J.; Behnke, M.; Chen, S.; Cruickshank, A. A.; Dick, L. R.; Grenier, L.; Klunder, J. M.; Ma, Y-T.; Plamondon, L.; Stein, R. L. Bioorg. Med. Chem. Lett. 1998, 8, 333. (f) Bailey, S.; Bolognese, B.; Buckle, D. R.; Faller, A.; Jackson, S.; Louis-Flamberg, P.; McCord, M.; Mayer, R. J.; Marshall, L. A.; Smith, D. G. Bioorg. Med. Chem. Lett. 1998, 8, 29-34. (g) Ohda, T.; Tsuchiya, N.; Nishimura, K.; Ikeda, E.; Wakayama J.; Takei, H. Bioorg. Med. Chem. Lett. 1996, 6, 543-546. (h) Kick, E. K.; Ellman, J. A. J. Med. Chem. 1995, 38, 1427-1430.
[5] Loh, Y. P.; Cawley, N. X.; Friedman, T. C.; Pu, L. P. In Aspartic Proteinases: Structure, Function, Biology, and Biomedical Implications; Takahashi, K., Ed.; Plenum Press: New York, 1995; pp 519-527.
[6] (a) Plunkett, M. J.; Ellman, J. A. J. Org. Chem. 1997, 62, 1885-2893. (b) Chenera, B.; Finkelstein, J. A.; Veber, D. F. J. Am. Chem. Soc. 1995, 117, 11999-12000. (c) Boehm, T. L.; Hollis Showlter, H. D. J. Org. Chem. 1996, 61, 6498-6499. (d) Woolard, F. X.; Paetsch, J.; Ellman, J. A. J. Org. Chem. 1997, 62, 6102-6103. (e) Hu, Y.; Porco, J. A. Jr.; Labadie, J. W.; Gooding, O. W.; Trost, B. M. J. Org. Chem. 1998, 63, 4518-4521.
[7] (a) Chan, T. H. Synthesis 1979, 761-786. (b) Han, Y.; Walker, S. D.; Young, R. N. Tetrahedron Lett. 1996, 37, 2703-2706.
[8] Schollkopf, U.; Groth, U.; Deng, C. Angew. Chem. Int. Ed. Engl. 1981, 20, 798-799.
[9] Miyarura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
[10] Farrall, M. J.; Fréchet, J. M. J. Org. Chem. 1976, 41, 3877. The 4-bromopolystyrene resin (50-100 mesh, 1.97 mmol/g resin, produced by co-polymerization of styrene, divinylbenzene (1%) and 4-bromostyrene) was purchased from Novabiochem Corp. (San Diego, CA).
[11] Bunin, B. A. The Combinatorial Index; Academic Press: San Diego, 1998.
[12] The cleavage conditions and loading levels for several of the polymer-bound compounds are as follows: 5, Br2 in CH2Cl2, 5 min, 0.40 mequiv/g; 6, Br2 in CH2Cl2, 5 min, 0.38 mequiv/g; 7a, UV/vis assay at 290 nm, 0.57 mequiv/g; 7c, 50% TFA in CH2Cl2, 24 h, 0.37 mequiv/g.
[13] It should be noted that during the deprotecton conditions for the Boc group (50% TFA in CH2Cl2 for 10 min), no protiodesilylated products were detected by 1H NMR spectral monitoring of the concentrated cleavage solution. This indicates that cleavage of the silicon-aryl bond occurs very slowly; therefore selective removal of the Boc-protecting group of amines can be achieved by treatment with acid (TFA) for a short period of time (10 min) without cleavage of the arylsilyl linkage.
[14] Ishiyama, T.; Abe, S.; Miyaura, N; Suzuki, A. Chem. Lett. 1992, 691.
[15] (a) Evans, D. A.; Britton, T. C.; Ellman, J. A.; Dorrow, R. L. J. Am. Chem. Soc. 1990, 112, 4001. (b) Evans, D. A.; Dorrow, R. L. Tetrahedron Lett. 1987, 28, 1123. (c) Evans, D. A.; Weber, A. E. J. Am. Chem. Soc. 1986, 108, 6757.
[16] Gademann, K.; Ernst, M.; Hoyer, D.; Seebach, D. Angew. Chem. Int. Ed. 1999, 38, 1223-1226.
[17] (a) Hintermann, T.; Seebach, D. Chimia 1997, 50, 244. (b) Seebach, D.; Abele, S.; Schreiber, J.; Martinoni, B.; Nussbaum, A. K.; Schild, H.; Schulz, H.; Hennecke, H.; Woessner, R.; Bitsch, F. Chimia 1998, 52, 734.
[18] (a) Matthew, J. L; Braun, C.; Guibourdenche, C.; Overhand, M.; Seebach, D. in Enantioselective Synthesis of a -Amino Acids, (Ed.; Juaristi, E.), Wiley, New York, 1996, pp. 105-126. (b) Cole D, C. Tetrahedron 1994, 50, 9517. (c) Kobayashi, S.; Ishitani, H.; Ueno, M. J. J. Am. Chem. Soc. 1998, 120, 431. (d) Ishitani, H.; Ueno, M. J.; Kobayashi, S. J. Am. Chem. Soc. 1997, 119, 7153.
[19] Cogan, D. A.; Liu, G.; Kim, K.; Backes, B. J.; Ellman, J. A. J. Am. Chem. Soc. 1998, 120, 8011-8019.
[20] Tang, T. P.; Ellman, J. A. J. Org. Chem. 1999, 64, 12-13.
[21] Dale, J, A.; Mosher, H. S. J. Am. Chem. Soc. 1973, 95, 512-519.
[22] Belofsky, G. N.; Jensen, P. R.; Fenical, W. Tetrahedron Lett. 1999, 40, 2913-2916.
[23] (a) Schmidt, U.; Lieberknecht, A.; Wild, J. Synthesis 1984, 53-60. (b) Schmidt, U.; Griesser, H.; Leitenberger, V.; Lieberknecht, A.; Mangold, R.; Meyer, R.; Riedl, B. Synthesis 1992, 487-490.
[24] (a) Burk, M. J.; Feaster, J. E.; Nugent, W. A.; Harlow, R. L. J. Am. Chem. Soc. 1993, 115, 10125-10138. (b) Burk, M. J.; Allen, J. G.; Kiesman, W. F. J. Am. Chem. Soc. 1998, 120, 657-663.
All comments on this poster should be sent by e-mail to (mailto:[email protected]) [email protected] with B0013 as the message subject of your e-mail.