
|
|
Fourth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-4), www.mdpi.org/ecsoc-4.htm, September 1-30, 2000
[C0006]
|
|
|
||
| . | . |
|
|
|
Unité de Phytopharmacie et Médiateurs Chimiques, INRA, Route de Saint-Cyr F-78026 Versailles Cedex, France Fax 01 30 83 31 19 E-mail [email protected] Received: 26 July 2000 / Uploaded: 9 August |
![]()
Introduction :
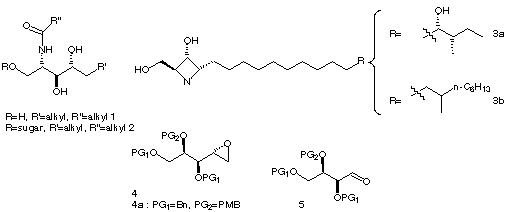 |
The scientific interest of the immunological properties of sphingolipids 11-6 and related glycosphingolipids 2 7 has recently increased on account of the role that they could play as therapeutic agents. Their antifongic properties have been the first studied 8. For number of them, immunomodulating activity as well as in vivo antitumoral activities have been reported 9-12. Some other compounds with original chemical structure have also been recently isolated from sponges 13-14
On the other hand, the original chemical structure and the promising
protein kinase C inhibitor activity of penarezidines 3a and penazetidines
3b
type of alkaloïds15-17 make them attracting
molecules in a therapeutic screening as well as in studies of other biological
phenomena including plant diseases involving cell growth disorders.
Although some syntheses18-28 of these
products have already been described in the litterature, the fact that
all these molecules were presenting a very similar feature consisting in
a terminal tetraheterosubstituted butyl subunit prompted us to design a
unifiying strategy involving a same synthetic versatile intermediate for
all these compounds elaborated from a carbohydrate synthon.
The more appropriate starting material, according to the well defined configurations of the stereogenic centers of these natural products was D-Glucose (Scheme 1a).
In all cases, the aliphatic side chain could be introduced in the final
steps of the synthesis by condensation of an appropriate organometallic
species on an epoxyde ring, the nitrogen atom beeing then introduce at
C-4 or C-2 (for sphingolipids) with inversion of configuration of this
carbon atom. For penazeridine and penazetidine synthesis, the azetidine
ring closure would be achieved by standart methodologies with inversion
of configuration at carbon C-2.
According to this retrosynthetic analysis, the target intermediate
of our strategy is thus epoxide 4 18 with
the appropriate protecting groups for the three remaining hydroxy groups.
A closely related strategy for the synthesis of penarezidine has been reported
earlier in the litterature and involves the use of the corresponding trihydroxy
butanal synthon 527 ; nevertheless,
the drawback of this strategy is that it requires, after condensation of
the organometallic species on the carbonyl group, an oxidation-reduction
sequence to obtain a good diastereomeric control of the configuration of
the hydroxy group formed during the alkylation step.
In rder to complmete the synthesis of the penaresidines from epoxide 4, three main routes can be used. First of all, route A described on Scheme 1b, which is the most similar to the works already described in the literature and which consists in the protection of the hydroxy group at C-2 before the introduction of the side chain through organometallic epoxide openning; the amino group is thereafter introduced at C-4 through substitution of a tosyl or mesyl group derived from the hydroxy group at C-4 generated during the epoxide openning. Nevertheless, in order to reduce the protection deprotection sequence of hydroxy or amino groups involved in the formation of the azetidine ring, one should consider the possibility of either introducing a leaving group at C-2 before the epoxide openning (Route A') or of keeping the hydroxy group at C-2 unprotected during this reaction and to manage the rest of the synthesis according to the presence of two free hydroxy groups in the molecule (Route B)
This poster deals with a new methodology for the synthesis of the polyhydroxylated azetidinic alkaloids encountered in the penaresidin and penazetidin family through route B described on Scheme 1b. The main feature of this synthesis lies in the absence of amine and/or hydroxyl protection/deprotection sequences usually encountered in the last steps of the synthesis devoted to the heterocycle ring closure.
Previous publications :
Beauhaire, J. ; Ducrot, P.-H. C. R. Acad. Sci. IIc 1999,
477-482.
Beauhaire, J. ; Ducrot, P.-H. Synth. Commun. 1998, 28,
2443-2456.
Results :
References : follow
this link
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with C0006 as the message subject of your e-mail.