
[C0019]
| Enzyme-Initiated Domino-(Cascade)-Reactions | |
W. Kroutil, S. F. Mayer and K. Faber
Department of Chemistry, Organic & Bioorganic Chemistry,
University of Graz, Heinrichstrasse 28, A-8010 Graz, Austria
Tel.: +43/(0)316/380-5332, Fax: +43/(0)316/380-9840, E-mail: [email protected]
URL: http://borgc185.kfunigraz.ac.at/
Received: 7 August 2000 / Uploaded: 10 August
1. Introduction
2. General
3.1 Enzyme-triggered Diels-Alder reactions
3.2 Domino-reactions initiated by enzymatically liberated
charge
3.3 Domino-reactions initiated by enzymatically liberated
nucleophile
4. Further reactions
5. Outlook
6. References and notes
Reactions proceeding through more than a single step in a concurrent fashion have been described in various context and different terms have been used to describe them, which renders some confusion. In order to provide a clearer picture, the following definition is used throughout this paper.
Type-I: Processes, where the starting material is undergoing a transformation via two (or more) reactions one after another in an inseparable fashion are denoted as 'domino-' or 'cascade-reactions'. The choice of words - domino, cascade - indicates that both individual reactions belong tightly together and are rather difficult to perform in a step-wise (independent) fashion. As a consequence, the intermediate between both steps is likely to be unstable and (often) eludes isolation and characterization.
Type-II: In contrast, 'sequential-' or 'tandem-reactions' are considered as two-step processes, which proceed in a consecutive fashion where each of the steps can be performed separately. Thus, it can be anticipated that the intermediate species will be a rather stable compound.
Type-I processes show a remarkable advantage: Despite of the fact, that the cascade-reaction is likely to proceed via a highly reactive (unstable) intermediate, which is prone to elude isolation and characterization, the final product can often be isolated in good yields, because decomposition of the reactive intermediate is largely avoided since it is transformed in the same instant as it appears. As a consequence, in ideal cases (when the velocity of both reactions is of the same order of magnitude) it does not occur in measurable concentrations. Unstable intermediates may constitute ionic or radical species and by convention, they are usually drawn in square brackets.
An impressive number of chemo-catalyzed cascade-reactions (especially those involving cyclizations) have been accomplished by using palladium [1-3]. Other types of typical cascade-reactions (although they have been denoted as 'tandem-reactions' in the respective publications) have been reviewed and classified according to their type of mechanism [4]. All of these reaction sequences were initiated by an organic or inorganic catalyst, or by thermal reactions. On the contrary, only few examples of cascade-reactions have been reported, where the initiation of the reaction cascade consisted of a biotransformation [5]. Taking advantage of the unparalleled diastereo- and enantio-specificity of enzymes, in many cases the reaction cascade was turned into an asymmetric fashion which furnished non-racemic product(s).
In this context, is should be stressed that the topic of this paper
involves the biotransformation of non-natural compounds (rather
than a substrate occurring in Nature). Thus, naturally occurring cascade-reactions
which are particularly common among terpenoid and steroid biosynthesis
pathways catalyzed by cyclases are out of scope of this review [6].
Emphasis is laid on domino-reactions, which are initiated via an
enzyme-catalyzed reaction, followed by a (spontaneous) subsequent chemical
reaction(s). These transformations have been aptly denoted as 'enzyme-initiated'
(or -'triggered') domino- (or cascade-)reactions.
Top of page
A survey of enzyme-triggered domino-reactions published to date reveal a common picture (Scheme 1): In a first step, the enzyme modifies a group ('trigger-group') within the starting material (e.g. via oxidation, transesterification of an alcohol, hydrolysis of an ester or epoxide, respectively), giving access to a reactive intermediate. This, for instance, may constitute a diene, or may bear a liberated negative charge, which can either push electrons into a p -electron-system or act as a nucleophile. Consequently, the intermediate thus formed immediately undergoes a subsequent 'domino'-reaction, which may consist of (i) a fragmentation, (ii) a rearrangement or (iii) a cyclization. The latter may involve a Diels-Alder reaction or an intramolecular SN2-reaction by the nucleophile liberated by the enzyme. Different classes of enzymatic domino-reactions and their types of follow-up reaction are summarized in Table 1.
Scheme 1: Principles of enzyme-initiated domino reactions.
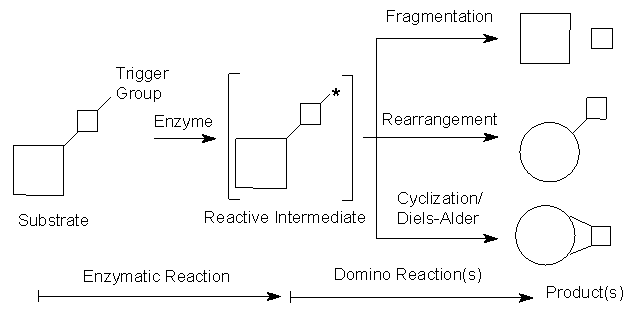
Table 1: Enzymatic transformation followed by domino
reaction(s).
|
Trigger-Reaction |
|
|
| phenol oxidation | diene formed | Diels-Alder |
| transesterification of alcohol | dienophile formed in kinetic resolution | intramolecular Diels-Alder |
| ester hydrolysis | electron-donating group liberated | retro-[2+2]cycloaddition/
fragmentation |
| ester hydrolysis | electron-donating group liberated | fragmentation |
| ester hydrolysis | electron-donating group liberated | rearrangement |
| ester hydrolysis | nucleophile liberated (-CO2-) | cyclization |
| ester hydrolysis | nucleophile liberated (-OH) | cyclization |
| epoxide hydrolysis | nucleophile liberated (-OH) | cyclization |
The first report on a deliberate combination of a biotransformation
with a chemical reaction to furnish a domino-sequence appeared as early
as 1981 [7], although Imai et al. may have observed
a bio-triggered cascade-reaction as an undesired side-reaction already
in 1976 by serendipity [8]. For the sake of clarity,
enzyme-initiated domino-reactions are grouped into the following subclasses:
(i) Diels-Alder reactions as domino-reaction, (ii) reactions initiated
by an enzymatically liberated charge, (iii) cyclizations involving enzymatically
generated nucleophiles and (iv) others.
Top of page
Diels-Alder reactions are frequently found in chemical cascade- or tandem-reactions [9], they are either employed as the first step [10] (e.g. combined with an aldol reaction [11]), or as the second step [12] (where the first step constitutes a Heck-reaction [13]). Another challenging strategy is the use of two consecutive Diels-Alder reactions during both steps. This was accomplished, either by using a specially designed (double) di-dienophile [14] or by employing a sequence consisting of a retro hetero-Diels-Alder followed by a second intramolecular Diels-Alder reaction [15]. In enzyme-triggered cascade-reactions containing a Diels-Alder reaction, the enzymatic step is always first.
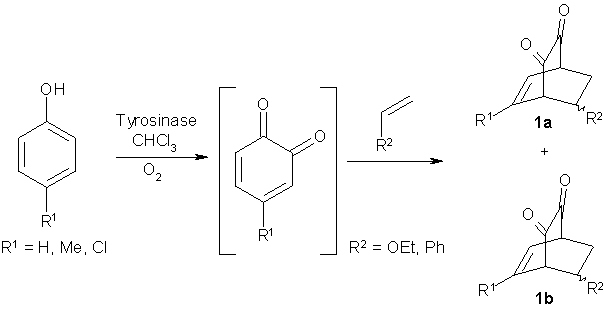
(i) Generation of a diene by enzymatic oxidation: In order to provide an unstable and highly reactive diene, substituted phenols were oxidized by immobilized tyrosinase at the expense of molecular oxygen to furnish the corresponding catechols and subsequently to o-quinones as intermediates (Scheme 2). Since the latter are prone to undergo polymerization, they cannot be isolated but were trapped in situ by reacting with a dienophile to form the bicyclic products of type 1 in up to 85% yield [16, 17]. The reactions were rather slow and took several hours up to three days. Although the products formed were essentially chiral, no asymmetric induction was noticed, since the cycloaddition reaction was of spontaneous nature and thus proceeded without the influence of the enzyme.
Scheme 2: Generation of diene by enzymatic oxidation followed by Diels-Alder reaction.
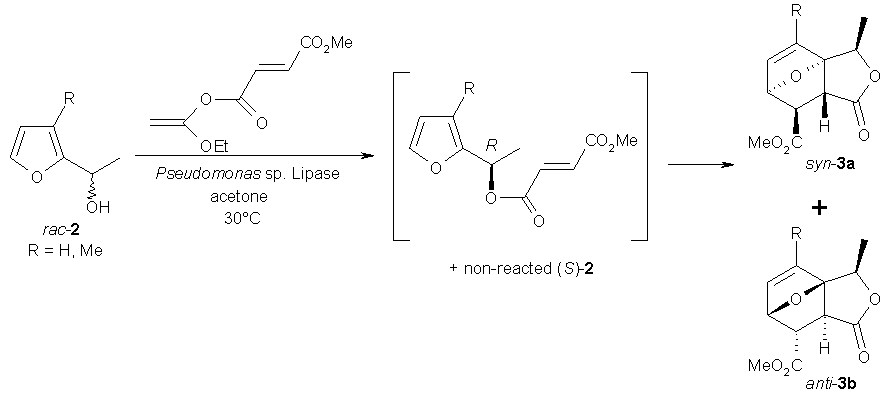
(ii) Generation of a dienophile via enzymatic kinetic resolution: An elegant case, where a Diel-Alder reaction was rendered in an asymmetric fashion leading to non-racemic product(s) is depicted in Scheme 3. In a first step, kinetic resolution of rac-furfuryl alcohol derivatives 2 was accomplished via acyl-transfer catalyzed by a lipase preparation (Toyobo-Lip, derived from Pseudomonas aeruginosa, immobilized on Hyflo Super-Cel) employing an enol ester (ethoxvinyl methyl fumarate) as acyl donor. In this way, two goals were achieved in a single step, i.e. diene and dienophile were linked onto each other in a single intermediate. The latter proceeded in an asymmetric manner via kinetic resolution of the sec-alcohol moiety. The second step constitutes of an intramolecular Diels-Alder reaction. This one pot-reaction rendered the final products 3 in low to moderate yields (18-43%) but in good e.e.´s (79-93%). As may be expected from the spontaneous (non-biocatalyzed) nature of the cycloaddition, the diastereoselectivity was shown to depend solely on the substituent R, ranging from low (R = H, d.e. ~25%) to excellent (R = Me, d.e. >99%) [18].
Scheme 3: Generation of dienophile via kinetic resolution, followed by Diels-Alder reaction.
| R |
|
|
| H |
|
|
| Me |
|
|
These types of reaction sequences have an enzyme-catalyzed hydrolytic starting step in common, during which an ester moiety is cleaved. As a consequence, an anion (e.g. a phenolate or carboxylate) was liberated. The latter does not participate itself in the subsequent reaction, but donates electrons into the molecule, thus enabling the domino reaction(s).
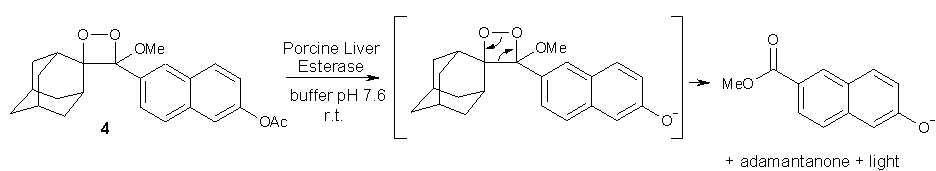
In the following reaction (Scheme 4), the acetate ester of a naphthol derivative containing a highly reactive 1,2-dioxetane moiety (4) was cleaved via hydrolysis employing porcine liver esterase liberating the free intermediate naphthol. The latter underwent an immediate fragmentation reaction which resulted in chemiluminescence [19].
Scheme 4: Induction of dioxetane fragmentation by ester hydrolysis.
A related reaction sequence was delineated in order to construct highly sophisticated protective groups for sensitive target molecules, such as bio-active (glyco- or lipo-)peptides. The protective groups could be selectively removed via enzyme-catalysis under mild conditions thus avoiding damage to the delicate bioactive target molecule. The protective group consisted of a phenol ester (R1-CO-) and a central aromatic moiety, bearing the target molecule (-R2) via an ester-, carbonate- or urethane linkage (Scheme 5). Depending on the reactive group within the target molecule, liberation occurs with (amine, alcohol) or without decarboxylation (carboxylic acid). The enzyme-trigger group (R1-CO-) may consist of a phenylacetate ester (which can be cleaved with absolute chemo-selectivity using Penicillin G acylase) or an aliphatic carboxyl ester (e.g. acetate, butanoate, octanoate), which is usually hydrolyzed by a lipase. The latter trigger-reaction liberates a phenolate anion, which (at an appropriate pH) leads to spontaneous fragmentation forming a p-quinomethane species going in hand with the expulsion of the target molecule or a reactive intermediate [a (hemi)carbonate or carbamic acid]. In the latter case, the intermediate undergoes further spontaneous decarboxylation to give the target alcohol or amine derivative [20, 21].
This strategy is also applicable for solid-phase synthesis, if the aromatic moiety building the scaffold is linked onto a macroscopic polymeric carrier via a spacer-arm, which renders an enzymatically labile anchoring group [22].
Scheme 5: Enzyme-triggered fragmentation of protective groups for amines, alcohols and carboxylic acids.

| R1 (Trigger group) | Enzyme | X | R1 (Target molecule) | |
| CH2Ph | Penicillin G Acylase | NH | Amine | |
| CH3, n-C3H7, n-C7H15 | Lipase | O | Alcohol | |
| CR2 | Carboxylix acid |
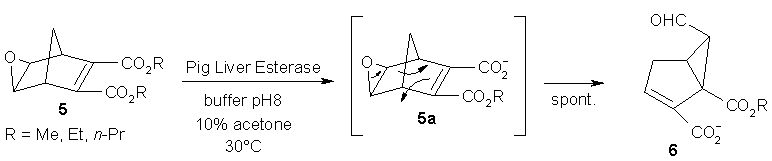
An unusual enzyme-triggered asymmetric rearrangement was observed by serendipity: When attempting to hydrolyze the bicyclic diester 5 in an asymmetric fashion using porcine liver esterase, the expected (chiral) monoester 5a was not obtained but the product turned out to consist of a bicyclo[3.1.0]hexane framework (Scheme 6). Detailed analysis revealed that the hemiester 5a was indeed formed, but immediately underwent Meinwald-rearrangement to furnish 6 in quantitative yield. Since the enzyme-triggered reaction was expected to proceed in an asymmetric fashion, the product was analyzed for its enantiomeric composition, which turned out to be a moderate 47 % e.e. for R being Me [23, 24].
Scheme 6: Asymmetric ester hydrolysis followed by Meinwald rearrangement.
Instead of undergoing a fragmentation or rearrangement reaction, the carboxylate or hydroxyl group formed during (enzymatic) ester- or epoxide-hydrolysis can also act as a nucleophile by attacking an electrophile during the cascade-reaction. Most interestingly, other strong nucleophiles (such as amines or thiols) were not yet investigated in this context. To date, the electrophile always consisted of an epoxide or a related species, such as halide [25]. Tosylate, or Michael-acceptors (such as enoates) etc. are waiting to be explored in this context.
Domino-reactions of this type started with the enzymatic hydrolysis of an ester or epoxide liberating a nucleophile (-CO2- or -OH), which opened an epoxide in an intramolecular SN2-reaction in the second step. Thus, the final product formed was a hydroxy-lactone (-CO2- as Nu) or tetrahydrofuran derivative (-OH as Nu).
Such a cascade-reaction was observed upon asymmetric hydrolysis of meso-epoxy-diester 7 using porcine liver esterase (PLE) [26] (Scheme 7). It was found that the more accessible (equatorial) carboxyl ester moiety was selectively hydrolyzed thus liberating a carboxylate anion, which in turn acted as a nucleophile by opening the epoxide moiety to furnish the corresponding hydroxy-g -lactone. In order to undergo lactone formation, the intermediate epoxy-carboxylate has to undergo a conformational change, which turns the second (remaining) axial ester moiety into the more accessible equatorial position. As a consequence, it could now be hydrolyzed as well by PLE and (1R,2S,4S,5S)-4-hydroxy-7-oxo-6-oxabicyclo[3.2.1]octane-2-carboxylic acid 8 was obtained as the final product in 96% e.e.
Scheme 7: g -Lactone formation initiated by enzymatically liberated Nu (-CO2-).

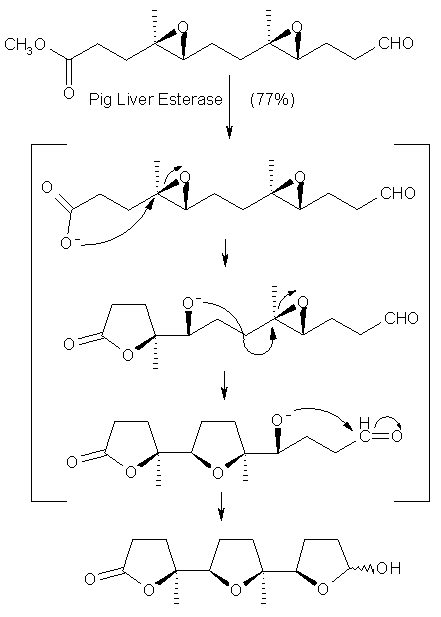
A related but more complex domino-reaction is depicted in Scheme 8. Again, the cascade started with an enzymatic hydrolysis of an ester liberating a nucleophile (-CO2-), which opened an epoxide to furnish the corresponding lactone together with a hydroxy moiety (-OH), which performed another (intramolecular) nucleophilic attack on the next epoxide to furnish tetrahydrofuran derivatives. As the end of this cascade, the final nucleophile (-OH) was trapped by forming a hemiacetal with an aldehyde bringing the cascade to a halt [27].
Scheme 8: Enzymatic liberation of Nu (-CO2-) followed by three-step SN2-cascade involving epoxy-groups.
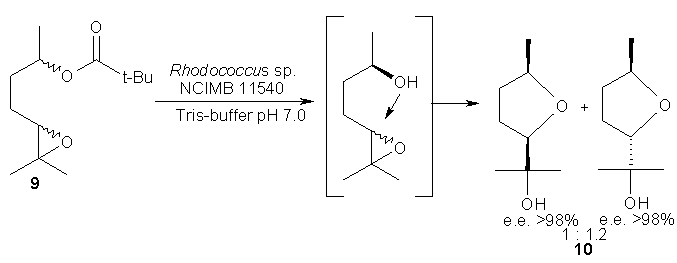
Instead of an enzymatically generated carboxylate anion, an alcohol group (derived from a biocatalyzed ester- or epoxide-hydrolysis) may as well serve as nucleophile to open an epoxy-moiety in a cascade-reaction. For instance, treatment of a diastereomeric mixture of (± )-epoxy ester 9 with a crude immobilized enzyme preparation (Novo SP 409 [28]) or whole lyophilized cells of Rhodococcus erythropolis NCIMB 11540 liberated the nucleophile (-OH) via kinetic resolution of the sec-alcohol moiety (e.e. >98%), which in turn opened the epoxide in an SN2-fashion thus furnishing the diastereomeric tetrahydrofuran derivatives 10 [29], which could be separated by conventional column chromatography.
Scheme 9: Cyclization initiated by enzymatically generated Nu (-OH) attacking an epoxide.
In all cases described above, the nucleophile acting during the cascade was liberated by the hydrolysis of an ester. In the following examples, the latter was furnished by an enzymatic hydrolysis of an epoxide to form the corresponding vic-diol.
Di-epoxide 11 was hydrolyzed by cytosolic epoxide hydrolase (cEH, from rat liver) in the first step to furnish a vic-diol, which reacted as a nucleophile in an intramolecular SN2-fashion with another epoxy-moiety in a related mode as described above [30]. Although some asymmetric induction might be expected in this cascade, the regio- and enantio-selectivity was not investigated in this paper. As a consequence, these results could have also been obtained via chemical catalysis. A related base-catalyzed cascade-reaction of optically active tri-epoxides has been published [31, 32] (Scheme 10).
Scheme 10: Liberation of Nu (-OH) via enzymatic hydrolysis of an epoxide, followed by intramolecular opening of another epoxy-moiety.

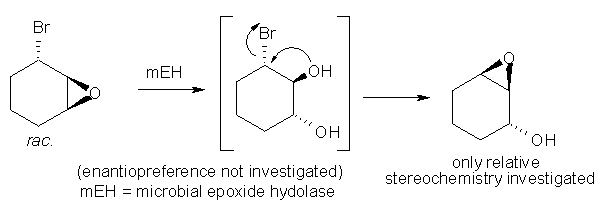
An alternative leaving group for the intramolecular SN2-reaction leading to ring closure represents a halogen in (± )-trans-3-bromo-1,2-epoxy-cyclohexene (Scheme 11). The nucleophile (-OH) obtained from enzymatic epoxide hydrolysis reacted with the bromo-moiety in a Payne-type rearrangement to form an epoxide [7].
Scheme 11: Liberation of Nu (-OH) via enzymatic hydrolysis of an epoxide, followed by intramolecular epoxide formation.
The first report on a bio-catalyzed cascade reaction (initiated by the hydrolysis of an epoxide) presumably dates back to 1976 [8]. It describes the microbial transformation of methyl epoxyfarnesate (12a) using the fungus Colletotrichum nicotianae via various pathways. One out of several products was identified as the tetrahydrofuran derivative 12b (Scheme 12). Based on the knowledge on the microbial transformation of terpenoid compounds available to date, it is reasonably to assume that the sequence proceeded via epoxidation of C=C bonds (by mono-oxygenases), followed by hydrolysis of the terminal epoxide, which led to spontaneous cyclization involving the internal epoxy-moiety.
Scheme 12: Microbial degradation of methyl farnesate involving a (putative) enzyme-triggered cyclization.

A careful survey of the literature reveals several transformations which cannot be accurately classified as they remain rather unclear in view of the catalyst(s) involved and/or the intermediates within the pathway.
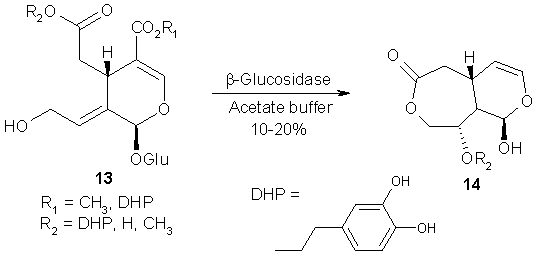
For instance, bio-hydrolysis of multifloroside 13 and its analogs afforded the rearrangement- product 14, which could not be obtained by using dilute acid (e.g. AcOH, HCl). As a consequence, it was assumed that the rearrangement might have been catalyzed by an enzyme. A possible mechanism still awaiting proof was proposed [33].
Scheme 13: Enzyme-triggered rearrangement of multifloroside (13).
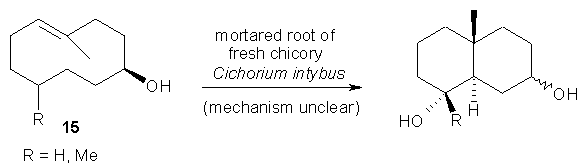
Finally, the cyclization of (E,E)-1,5-cyclodecadienol 15 by mortared root of fresh chicory (Cichorium intybus) constitutes an example for an enzyme-triggered cyclization-reaction of a non-natural compound. In this case the biocatalyst involved as well as the mechanism are still unknown [34, 35].
Scheme 14: Cyclization catalyzed by a preparation of fresh chicory.
The application of enzyme-triggered cascade-reactions for the transformation of non-natural compounds offers two distinct advantages: Firstly, the final product can often be obtained in good yield despite the fact that the reaction sequence involves several steps through highly reactive and thus ostensibly unstable intermediate species. Secondly, based on the exquisite diastereo- and enantio-selectivities of biocatalysts, pathways may often be conducted in an asymmetric fashion, thus non-racemic products are obtained.
Until now, in all cascade-reactions involving a biotransformation, the
biocatalyst was employed in the first step. This is likely to be the most
economic strategy, since asymmetry is introduced at the very beginning
of the cascade. Since the design of enzyme-triggered cascade-reactions
is not trivial and (out of neccessity) will always involve rather complex
molecules, these processes are unlikely to become a general tool but they
definetely offer an intelligent option for asymmetric syntheses. Given
the knowledge to date, this field as a whole is almost non-exploited and
(to our belief) its potential is grossly underestimated. Detailed studies
on this topic are carried out in our laboratories.
Top of page
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with C0019 as the message subject of your e-mail.