|
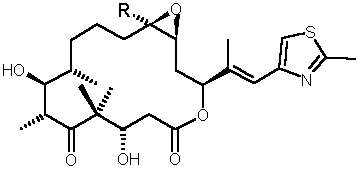
Figure 1: Structures of Epothilones A (R = H) and B (R = CH3) |
[C0024]
Synthetic Epothilone Analogs with Modifications in the Northern Hemisphere and the Heterocyclic Side-Chain - Synthesis and Biological Evaluation
Nicole End, Guido Bold, Giorgio Caravatti, Markus Wartmann, and Karl-Heinz Altmann*Novartis Pharma AG, TA Oncology Research,
WKL-136.4.21, CH-4002 Basel, Switzerland
Fax: ++41-61-6966246. E-mail: [email protected]
Received: 7 August 2000 / Uploaded: 10 August
Introduction
Epothilones A and B
I (Fig. 1) are naturally occurring 16-membered macrolides, which are produced by the myxobacterium Sorangium cellulosum. [1] Although these compounds do not share any obvious structural similarities with the prominent anticancer drug paclitaxel (Taxol�), they exhibit a similar biological profile in vitro, including the ability to inhibit microtubule depolymerization and to induce apoptosis in human cancer cell lines with sub-nM IC50�s. [2] [3] Epothilone B is a 3-20-fold more potent inhibitor of human cancer cell growth than paclitaxel and unlike paclitaxel is also effective against multidrug-resistant cell lines. Epothilone B has demonstrated potent in vivo antitumor activity [3] in and is currently undergoing Phase I clinical trials by Novartis.
|
Figure 1: Structures of Epothilones A (R = H) and B (R = CH3) |
The biological profile of natural epothilones has triggered substantial synthetic efforts directed at the exploration of the SAR of this class of natural products and the discovery of analogs with an improved pharmacological profile. [4] This has led to a rather comprehensive understanding of the SAR of epothilones within an unusually short (given the complexity of these molecules) time period after the disclosure of their absolute stereochemistry in mid-1996. [5] Among others, three of the most relevant SAR features include the facts that (i) the presence of a C12-C13 epoxide moiety is not absolutely required for potent microtubule stabilization and profound antiproliferative activity (the epoxide may be replaced by a simple C12-C13 double bond), (ii) analogs incorporating a trans epoxide or trans olefin structure at C12/C13 appear to be almost equipotent with the corresponding cis isomers, and (iii) the replacement of the thiazole ring either by an oxazole or various pyridine [6] moieties is well tolerated. Within the context of these general aspects of the epothilone SAR this paper addresses three specific questions each of which is related to one of the above types of modifications:
|
|
|
|
|
Results and Discussion
1. Retrosynthesis
The retrosynthetic analysis for target structures 1 - 3 is summarized in Schemes 1A (1) and 1B (2, 3). In all three cases the key steps for construction of the macrocyclic skeleton involve Yamaguchi macrolactonization, the build-up of the requisite seco-acid through aldol reaction between the C7-C15 aldehyde and the dianion of the O-protected C1-C6
b-hyroxy acid fragment, and the assembly of the C7-C15 aldehyde through the appropriate type of Pd(0)-catalyzed coupling reaction. In the case of 2a/b macrolactonization and deprotection were to be followed by stereoselective epoxidation of the trans C12-C13 double bond. This approach in essence represents a combination of the strategies developed by Schinzer et al. [11] and Nicolaou et al. [4a] for the synthesis of the natural products.| Scheme 1 |
| A |
 |
| B |
 |
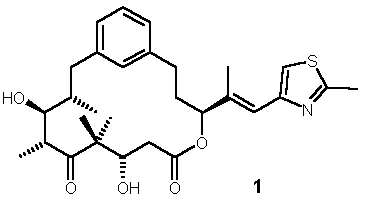
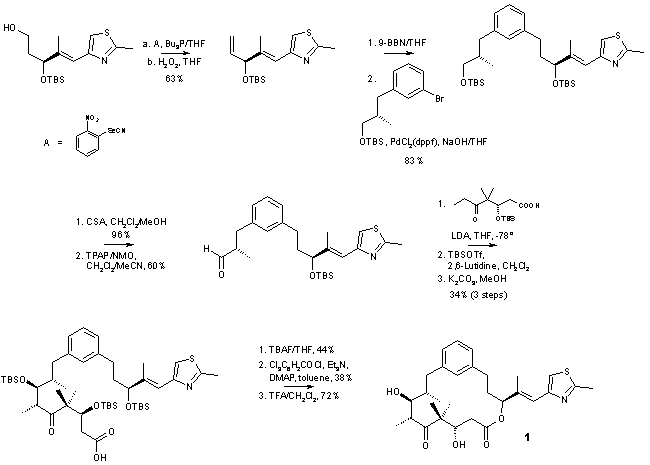
2. Synthesis of Conformationally Constrained Epothilone Analog 1
The synthesis of epothilone analog 1 is summarized in Schemes 2 and 3. The protected C7-C12 fragment (red structure in Scheme 1, R = TBS) was obtained from commercially available 3-bromocinnamic acid as the starting material, using Evans chemistry to establish the chiral center at C8 (epothilone numbering, Scheme 2)) . The critical junction between fragments C7-C12 and C13-C15 was achieved through alkyl Suzuki coupling between the C7-13 aryl bromide and a C13-C15 terminal olefin in excellent 83% yield. Transformation of this coupling product to the aldehyde and subsequent aldol reaction with the dianion of the O-protected
b-hydroxy acid comprising the C1-C6 fragment of epothilones yielded the desired anti-Felkin syn aldol product with 5:1 selectivity. The b-hydroxy acid (green structure in Scheme 1, R = TBS) was prepared as suggested by de Brabander et al. [12] However, it should be noted that contrary to what is reported in ref. [12], the preparation of the desired (3S)-enantiomer requires the use of the (2R)-bornane-10,2-sultam as chiral auxiliary. The synthesis was completed by Yamaguchi macrolactonization, which proceeded in moderate yield, followed by deprotection with TFA/CH2Cl2.Scheme 2: Epothilone Analog 1 - Synthesis of the C7-C12 Fragment
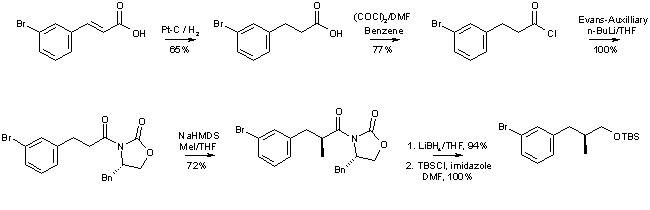
Scheme 3: Epothilone Analog 1 - Synthesis of the C13-C15 Fragment and Construction of the Macrocycle
3. Synthesis of Trans-Epothilones A 2a/b
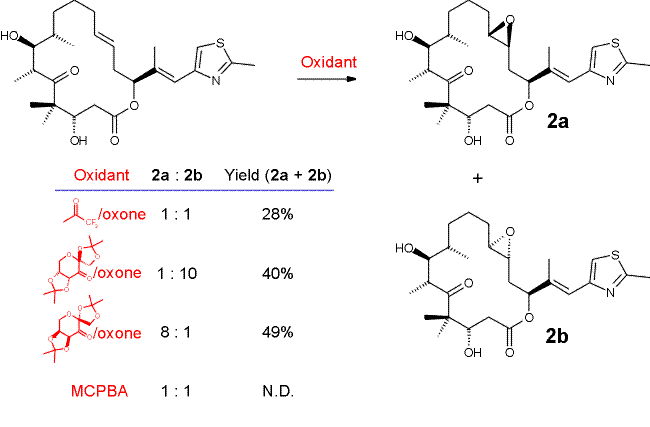
The stereoselective synthesis of trans-epothilones A is summarized in Schemes 4 � 7. The strategies for the preparation of the C7-C11 (Scheme 4) and C12-C15 (Scheme 5) fragments are closely related to those devised by Schinzer et al. [9] and either involve Evans chemistry or a chiral pool starting material ((S)-malic acid; fragment C12-C15, Scheme 5) to establish the required chiral centers. The coupling of fragments C7-C11 and C12-C15 in this case was achieved through Pd(0)-catalyzed reaction between the C12-C15 trans vinyl iodide and the zincate derived from the C7-C11 alkyl iodide in 64% yield (Scheme 6). The most critical step in the preparation of trans-epothilones A proved to be the chemo- and stereoselective epoxidation of the C12-C13 trans double bond, which was finally accomplished using fructose derived epoxidation catalysts developed by Shi [13] (Scheme 7). The stereochemistry of the epoxidation products was unambiguously established by X-ray crystallographic analysis of compound 2a.
Scheme 4: Trans-Epothilones A - Synthesis of the C7-C11 Fragment
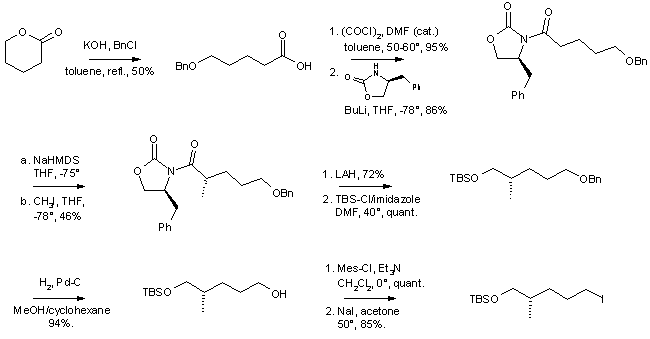
Scheme 5: Trans Epothilones A - Synthesis of the C12-C15 Fragment

Scheme 6: Trans-Epothilones A - Synthesis of the Olefin Precursor

Scheme 7: Trans-Epothilones A � Epoxidation Selectivity
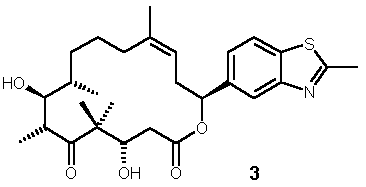
4. Synthesis of Side-chain modified Deoxyepothilone B Analog 3
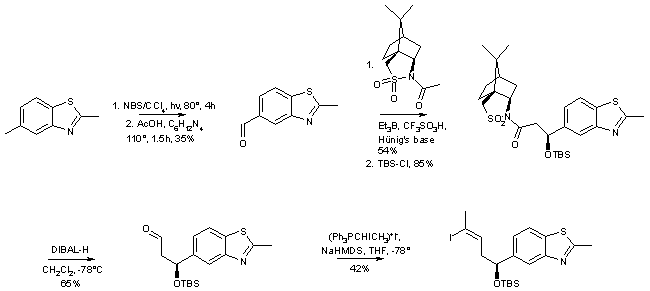
The synthesis of side-chain modified deoxyepothilone B analog 3 is summarized in Schemes 8 and 9. The retrosynthetic analysis outlined in Scheme 1 called for a C12-C15 cis-vinyl iodide as one of the coupling components for the construction of the macrocycle. This compound was obtained through aldol reaction between 2-methylbenzothiazole-5-carbaldehyde and (2R)-N-acetylbornane-10,2-sultam (ca. 4/1 selectivity), followed by O-protection, reduction to the aldehyde and Wittig-olefination [14] (Scheme 8). The following construction of the macrocycle mirrors the chemistry employed for the synthesis of 2a/b, except that a significantly lower yield was obtained in the macrolactonization step, which was accompanied by the formation of significant amounts of the cyclic dimer (Scheme 9).
Scheme 8: Deoxyepothilone B analog 3 � Synthesis of the C12-C15 Vinyl Iodide
Scheme 9: Deoxyepothilone B analog 3 � Construction of the macrocycle

5. Biological Activity
Epothilone analogs 1, 2a/b, and 3 were assessed for their ability to induce tubulin polymerization in vitro (as a measure for their microtubule stabilization potential) and to inhibit the growth of human cancer cells, either in a drug-sensitive or a drug-resistant background. The results of these studies are summarized in Table 1.
Table 1: Biological Activity of Epothilone Analogs.
Growth Inhibition (IC50 [nM]) b |
|||
Epothilone analog |
Tubulin Polymerization (%) a |
KB-31 c |
KB-8511 d |
1 |
< 10 |
> 1000 |
> 1000 |
2a |
85 |
1.00 |
0.87 |
2b |
< 10 |
523 |
305 |
3 |
79 |
0.45 |
0.23 |
Epothilone A |
65 |
2.15 |
1.91 |
Epothilone B |
85 |
0.20 |
0.20 |
Deoxyepothilone B |
93 |
2.70 |
1.44 |
Paclitaxel (Taxol�) |
44 |
2.84 |
615 |
aInduction of tubulin polymerization at 2
mM compound concentration relative to the effect of 25 mM Epothilone B, which is defined as the 100% level. bCancer Cell growth inhibition was assessed after a 72h exposure period. cHuman epidermoid cancer cell line, which is sensitive to growth inhibition by most common anticancer agents. dP-glycoprotein (P-gp)-overexpressing subline of the KB-31 parental line, which is resistant to treatment with paclitaxel.Epothilone analog 1 proved to be devoid of any appreciable biological activity. This may either indicate that our pharmacophore model does not appropriately reflect the bioactive conformation of epothilones; alternatively, the additional steric bulk associated with the phenylene moiety could conceivably more than outweigh any possible activity gain arising even from proper conformational stabilization. With regard to trans-epothilones A the data in Table 1 clearly demonstrate that 2a is at least equipotent with, if not more potent than epothilone A itself, while its diastereoisomer 2b is 500-fold less potent. Likewise, 3 exhibits significantly higher biological activity than its parent compound deoxyepothilone B. Based on these in vitro data 2a as well 3 represent promising candidates for further profiling in vivo.
6. Acknowledgement
The skilled technical assistance by K. Hauenstein, W. Vetterli, J. Koppler, and M. Lartigot is gratefully acknowledged. N. van Campenhout and P. Furet are thanked for the development of the epothilone pharmacophore model.
7. References
[1] a) G. Hofle, N. Bedorf, K. Gerth, H. Reichenbach, German patent disclosure DE 4138042, May 5, 1993 (Priority Nov. 19, 1991). b) K. Gerth, N. Bedorf, G. Hofle, H. Irschik, H. Reichenbach, J. Antibiotics 1996, 49, 560-564.
[2] a.) D. M. Bollag, P. A. McQueney, J. Zhu, O. Hensens, L. Koupal, J. Liesch, M. Goetz, E. Lazarides, C. A. Woods, Cancer Res. 1995, 55, 2325-2333. b.) R. J. Kowalski, P. Giannakakou, E. Hamel, J. Biol. Chem. 1997, 272, 2534-2541.
[3] K.-H. Altmann, M. Wartmann, T. O�Reilly, Biochimica et Biophysica Acta 2000, 1470, M79-M91.
[4] For reviews, cf.: a) K. C. Nicolaou, F. Roschangar, D. Vourloumis, Angew. Chem. 1998, 110, 2120-2153; Angew. Chem. Int. Ed. Engl. 1998, 37, 2014-2045. b) C. R. Harris, S. J. Danishefsky, J. Org. Chem. 1999, 64, 8434-8456.
[5] G. Hofle, N. Bedorf, H. Steinmetz, D. Schomberg, K. Gerth, H. Reichenbach, Angew. Chem. 1996, 108, 1671-1673; Angew. Chem. Int. Ed. Engl. 1996, 35, 1567-1569.
[6] K. C. Nicolaou, R. Scarpelli, B. Bollbuck, B. Werschkun, M. Manuela, A. Pereira, M. Wartmann, K.-H. Altmann, D. Zaharevitz, R. Gussio, P. Giannakakou, Chemistry & Biology 2000, in press.
[7] N. van Campenhout, P. Furet, unpublished results.
[8] K. C. Nicolaou, Y. He, D. Vourloumis, H. Vallberg, F. Roschangar, F. Sarabia, S. Ninkovic, Z. Yang, J. I. Trujillo, J. Am. Chem. Soc. 1997, 119, 7960-7973.
[9] K. C. Nicolaou, D. Vourloumis, T. Li, J. Pastor, N. Winssinger, Y. He, S. Ninkovic, F. Sarabia, H. Vallberg, F. Roschangar, N. P. King, M. R. V. Finlay, P. Giannakakou, P. Verdier-Pinard, E. Hamel, Angew. Chem., Int. Ed. 1997, 36, 2097-2103.
[10] X-Ray crystal strucutre of epothilone A: G. Rihs, unpublished data.
[11] D. Schinzer, A. Bauer, J. Schieber, Chem.-Eur. J. 1999, 5, 2492-2500.
[12] J. De Brabander, S. Rosset, G. Bernardinelli, Synlett, 1997, 824-826.
[13] Y. Tu, Z.-X. Wang, Y. Shi, Yian, J. Am. Chem. Soc. 1996, 118, 9806-9807.
[14] J. Chen, T. Wang, K. Zhao, Tetrahedron Lett. 1994, 35, 2827-2828.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with C0024 as the message subject of your e-mail.