
[C0027]
A New Binding Motif for Amidinium Cations – Strong Binding of Amidinium Salts with m-Xylylene Bisphosphonate Moieties
Thomas Grawe,a Thomas Schrader*b
aInstitut für Organische Chemie und
Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf,
Universitätsstr. 1, D-40225 Düsseldorf, Germany
bFachbereich Chemie, Philipps-Universität Marburg,
Hans-Meerwein-Straße, D-35032 Marburg, Germany
E-mail: [email protected]
Received: 7 August 2000 / Uploaded: 12 August
Introduction
In contrast to the multitude of different binding motifs for the molecular recognition of ammonium cations very few efficient guanidinium receptors have been reported and even less amidinium hosts [1]. The latter is surprising in view of important role of the amidinium functionality in drugs targeting binding pockets for the arginine sidechain etc. The challenge, however, is quite clear: One has to surround the amidinium group in a halfmoon-like array with at least four hydrogen bond acceptors, which are ideally preoriented for maximum electrostatic as well as hydrogen bond interactions.
Table 1. Association constants (K
1:1) [M-1] from NMR titrations with 1 and 2 in DMSO at 20°C.[a]|
Guanidinium or amidinium derivative |
Receptor molecule |
(K1:1)[M-1] |
DG [kcal/mol] |
|
N- a-tosylarginine methyl ester |
1 |
86000 ± 28% |
6.7 |
|
1,1-Dimethylguanidine |
1 |
36000 ± 38% |
6.2 |
|
Methylguanidine |
1 |
25000 ± 35% |
6.0 |
|
N- a-tosylarginine methyl ester |
2 |
21800 ± 13% |
5.8 |
|
1,1-Dimethylguanidine |
2 |
6600 ± 14% |
5.1 |
|
Methylguanidine |
2 |
5600 ± 39% |
5.0 |
|
Acetamidine (2:1-complex) |
2 |
1000 ± 20% |
4.0 |
a Due to the strongly hygroscopic character of both titration partners the d6-DMSO solution contained ~0.1% of water. Errors in Ka are standard deviations and were calculated to be ± 6-39%.
Recently we introduced molecular tweezers which are highly selective for alkylguanidinium cations (e.g., arginine derivatives) (Fig. 1) [2]. When we checked their binding behaviour against higher substituted guanidines, we were surprised: Although the double substitution in N,N-dimethyl guanidinium salts renders their molecular recognition by bisphosphonate tweezers in the ordinary binding mode (A) impossible, they were indeed bound even more tightly than monomethyl guanidinium cations (Table 1). Both, the parent compound m-xylylene bisphosphonate 1 as well as the diphenyl ether tweezer 2 showed this interesting effect. After extensive molecular modeling [3] and screening of X-ray structures of guanidinium salts we now propose a new binding mode (B), which is again a chelate, albeit with a slighty (60°) rotated guest moiety (Fig. 1). Here all hydrogen bonds are kinked, but the mutual phosphonate repulsion is weakened (d1<d2, Fig. 2).
Fig. 1. Molecular tweezers 1 and 2.

Fig. 2. A) Ordinary binding mode of alkyl guanidinium cations by bisphosphonate 1. B) Alternative molecular recognition pattern for N,N-dialkyl guanidinium cations.
Although in general arrangement A is found ubiquitiously, some X-ray structures document the existence of binding pattern B [4]. This unusual conformation was also formed when we complexed 1 with N/C-protected arginine derivatives. At concentrations below 0.7 mM all arginine proton signals shifted according to a 1:1-binding isotherm – except for the guanidinium-NH, which remains untouched. We conclude, that again arrangement B is formed preferentially. Force-field calculations demonstrate that this is favorable for a folding of the amino acid side chain which brings the acyl- or tosyl-NH-functionality in close proximity to one of the phosphonate anions (Fig. 3) [2]. The strong downfield-shift of the respective NH-proton indicates the formation of this additional intermolecular hydrogen bond, which in a cooperative effect increases the overall binding constant to 86.000 M-1(Table 1).
If, however, for a strong recogonition of guanidinium cations by bisphosphonates only 4 hydrogen bonds of two adjacent NH2-groups are necessary, could the third nitrogen atom be replaced by carbon or oxygen? In this case a new receptor structure for amidines or even urea would have been found! (Fig. 4) Molecular recognition of amidines is especially interesting because of the high number of medicinally important bisamidine derivatives, which are currently used against the AIDS-associated pathogen Pneumocystis carinii. The related pneumonia occurs with about 80% of all patients. The above-mentioned drugs bind to AT-rich sequences in this pathogen’s genome and thus prevent its replication [5]. Urea has always been problematic during dialysis; a strong and selective recognition of urea in water by an artificial receptor molecule could solve this problem [6].
a) |
|
Fig. 3. a) Energy-minimized complex from a-N-tosyl arginine methyl ester and bisphosphonate receptor molecule 1; b) molecular mechanics calculation for the related complex between acetamidinium cation and 1.
Fig. 4 demonstrates how the same molecular arrangement could be used not only for recognition of dialkyl guanidines, but also for amidines and urea. To check our hypothesis we titrated acetamidinium chloride with the bisphosphonate candidates 1 and 2 (Fig. 5) [7]. The binding curves are presented in Fig. 5 and 6.

Fig. 4. Molecular recognition of amidines and urea according to the same binding pattern as of N,N-dialkyl guanidines?
Complex formation with tweezer 2 produces a smooth curvature for all NMR signals; however, only the inner NHi-proton signal is strongly shifted downfields, whereas the outer NHo-proton signal even migrates highfields. This is typical for the classical amidinium binding pattern with carboxylates or phosphonates [8]. The nonlinear curve fitting confirms the expected 1:2-complex stoichiometry (Fig. 5) and the value for the calculated association constant Ka ~ 1000 M-1 lies in the usual range [7].

Fig. 5. Chemically induced shift (CIS) of the signals of acetamidine hydrochloride during the NMR titration with increasing amounts of the bisphosphonate tweezer 2 in DMSO-d6 at 20°C.
The binding curve for m-xylylene bisphosphonate 1, however, differs drastically from the classical behaviour (Fig. 6): The CIS for the inner amidinium protons NHi first rises steeply, then beyond 0.5 equivalents flattens and at 1 equivalent almost reaches saturation. The respective NMR signal for the outer protons NHo remains unchanged up to 0.5 equivalents, then starts to shift downfields, but reaches saturation not before ~ 1.5 equivalents of receptor molecule. This anormal behaviour is easily explained with the assumption, that up to 0.5 equivalents of receptor molecule (here just one phosphonate group binds one amidinium cation) the classical binding pattern dominates (Fig. 6).
 |
 |
Fig. 6. CIS for NMR signals of a) acetamidine hydrochloride und b) benzamidine hydrochloride during the NMR titration with increasing amounts of m-xylylene bisphosphonate 1 in DMSO at 20°C.
As soon as an excess of bisphosphonate molecules appears, these begin with formation of the chelate complex involving also the outer NHo protons in hydrogen bonds. At ~ 1.5 equivalents only this chelate-type 1:1-complex is present (Fig. 6). The alkyl rest produces a medium signal which comes already very close to a 1:1-binding isotherm. Force-field calculations offer an explanation for the markedly lower downfield shift of NHo compared to NHi. For steric reasons only one of both NHo-protons seems to be able to form a direct hydrogen bond with a phosphonate anion (Fig. 3.b). A similar titration with benzamidine gave exactly the same curve picture, but with a much larger downfield shift for NHo of 0.3 ppm.
We attempted to reach a uniform 1:1 complex stoichiometry at a 5 times lower concentration (isolated particles); although the sigmoidal character of the resulting binding curve decreased, it was still retained. To obtain reliable 1:1-binding constants we finally performed dilution experiments in even more diluted solutions with a twofold excess of receptor molecules (2 equivalents per amidine) to be sure that all molecules were complexed in the 1:1-binding mode [9]. Now all curves followed an almost ideal 1:1-binding isotherm (Fig. 7); the association constants of various amidinium chlorides were again calculated by nonlinear regression and are listed in Table 2.

Fig. 7. CIS of proton NMR signals of acetamidine hydrochloride during a dilution experiment with a mixture of 2 equivs. of bisphosphonate 1 and 1 equiv. of amidinium chloride in DMSO-d6 at 20°C.
All association constants lie two orders of magnitude higher than the classical amidinium-phosph(on)ate complexes (105 M-1 vs. 103 M-1); thus, the amidinium-bisphosphonate complexes are among the most stable assemblies of amidines with an artificial receptor molecule. Bell developed a concave, highly preorganized receptor molecule based on anullated pyridines which also binds benzamidine very efficiently (Ka in 10 % methanolic dichloromethane ~ 107 M-1; Fig. 8) [10]. However, this molecule is only accessible through a multistep-synthesis as opposed to the simple m-xylylene bisphosphonate 1.

Fig. 8. Molecular recognition of benzamidine by a highly preorganized artificial receptor molecule (Bell).
Comparison of the association constants for various substituted benzamidines shows that these remarkably correlate with the electronic character of the substituents. Thus a fine-tuning of predictable binding-constants depending on the respective Hammet constants should be possible [11]. A similar correlation between binding strength and electronic character of the substituents has also been discovered when various benzamidines were docked onto trypsine and thrombine in the binding pocket for the arginine side chain [12]. Even these very small substrates („needles“) are potent inhibitors with Ki-values in the micromolar range.
Table 2. Association constants (K
1:1) [M-1] from NMR-dilution experiments with 1 in DMSO at 20°C.[a]|
Amidine hydrochloride |
(K1:1)[M-1] |
DG [kcal/mol] |
|
p -Methoxybenzamidine |
76000 ± 13% |
6.6 |
|
Acetamidine |
103000 ± 15% |
6.8 |
|
Benzamidine |
120000 ± 17% |
6.9 |
|
p -Amidinobenzamide |
140000 ± 30% |
7.0 |
|
m -Nitrobenzamidine |
250000 ± 20% |
7.3 |
a Due to the strongly hygroscopic character of both titration partners the d6-DMSO solution contained ~0.1% of water. Errors in Ka are standard deviations and were calculated to be ± 13-30%.
Bisamidines play a prominent role in AIDS therapy: The drug pentamidine binds to DNA according to several detailed binding studies not intercalating, but in the minor groove along the phosphodiester backbone [13]. For the electrostatic attraction on the amidinium end groups the center of maximum negative charges is used, which resides deeply in the interior of the double helix, i.e. shortly above the nucleic bases. The rest of the guest molecule forms a wide curve with a perfect fit into the minor groove, a conformation which is also the thermodynamically most stable one (Fig. 9) [14]. All the other medicinally active bisamidines are bound similarly. We feel that this environment can be effectively imitated with a receptor molecule of the correct shape, containing two bisphosphonate moieties. Work towards this goal is underway in our laboratory.
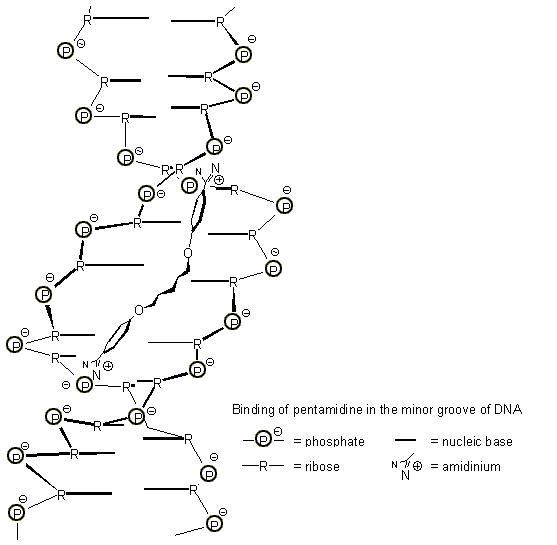
Fig. 9. Binding of pentamidine in the minor groove of DNA (according to ref. 14).
Conclusions and Outlook
We conclude that chelate complexes of amidinium cations with benzylic bisphosphonates represent a new highly efficient binding motif for this substance class. Since benzylic bisphosphonates are readily accessible by standard methods with inexpensive reagents, this moiety can easily be incorporated in new materials for the affinity chromatography used for drug purification or isolation of precious natural products. Another potential application lies in the area of molecular imprinting: we suggest that polymerizable bisphosphonates constitute a new prototype of binding monomers with promising features for the selective recognition of amidinium substrates in the cavities of macroporous highly crosslinked imprinted copolymers.
Experimental Section:
General Methods: Dimethyl sulfoxide (Merck) was purchased in 99.8% purity.
Bis(tetrabutylammonium) m-xylylene-P,P'-dimethyl diphosphonate 1: m-Xylylene di-phosphonic acid tetramethyl ester was treated with 2.0 eq. of aqueous tetrabutylammonium hydroxide and heated to reflux for 1 week. After evaporation to dryness, the crude product was extracted with chloroform, dried over magnesium sulfate, filtered and again evaporated to dryness. The receptor was further dried at 10-3 mbar over P2O5. Yield: 92%. Mp.: ~40°C. 1H NMR (300 MHz, [D6]DMSO): d = 0.94 (t, 24H, J = 7.3 Hz), 1.31 (tq, 16H, J = 7.3 Hz), 1.57 (m, 16H), 2.53 (d, 4H, J = 18.5 Hz), 3.18 (m, 16H), 3.21 (d, 6H, J = 9.8 Hz), 6.92-6.95 (m, 4 H). 13C NMR (75 MHz, CDCl3): d = 13.8 (s, 8 C), 19.7 (s, 8 C), 24.1 (s, 8 C), 35.7 (d, J = 124.0 Hz, 2 C), 51.61 (m, 2 C), 58.46 (m, 8 C), 126.5 / 126.9 / 131.6 (m, 4CH), 137.7 (m, 2C). 31P NMR: d = 16.48 (s). Elemental analysis: calcd. or C42H86N2O6P2 x 3H2O: C 60.69, H 11.16, N 3.37, found: C 60.54, H 11.33, N 3.35.
Bis[p-(dimethoxyphosphorylmethyl)phenyl]ether (starting material for molecular tweezer 2): Di-p-tolylether was converted to the respective dibromide with two molar equivalents of NBS and catalytic amounts of AIBN in dry tetrachloromethane by heating the reaction mixtures to 80°C. After recrystallization the dibromide was heated for 3 h to 140°C with two molar equivalents of trimethylphosphite. Then reasts of trimethylphosphite and methylphosphonic acid dimethylester were evaporated in vacuo. Chromatographic purification over silica gel (ethyl acetate / methanol = 10:1) affords an analytically pure colourless oil which crystallizes during one day. Yield: 26% overall; M.p. 86-7°C; 1H NMR (300 MHz, CDCl3): d = 3.12 (d, 4H, J = 21 Hz, CH2P), 3.63 (d, 6H, J = 10.5 Hz, CH3O), 6.73 (d, 4 Harom., J = 8 Hz), 7.18 (dd, 4 Harom., J = 8/2 Hz)(AA´BB´-system). 31P NMR (CDCl3): d = 29.48 (s). Elemental analysis: calcd. for C18H24O7P2: C 52.18, H 5.84, found C 52.20, H 5.75.
Bis[(tetrabutylammonium)-p-(methoxyoxyphosphorylmethyl)phenyl]-ether 2: The tetra-methyl phosphonates were heated to 120°C for ca. one week with exactly two molar equivalents of a aqueous tetrabutylammonium hydroxide solution (1.5M). After evaporation to dryness and extraction with dry chloroform the solution was dried over magnesium sulfate, filtered and again evaporated to dryness. The oily product was purified from rests of the solvent and traces of water by stirring at 60°C in vacuo. After ca. one week the product crystallized slowly. Even after this drying procedure the receptor contained exactly one molecule of water per phosphonate group; obviously water is bound very tightly to this host (cf. elemental analysis). Yield: 95%; M.p. 50-1°C; 1H NMR (300 MHz, CDCl3): d = 0.99 (t, 24H, J = 7.2 Hz, CH3), 1.40 (tq, 16H, J = 7.2 Hz, 8 CH2), 1.60 (m, 16H, 8 CH2), 2.97 (d, 4H, J = 19.8 Hz, CH2P), 3.26 (m, 16H, CH2N), 3.49 (d, 6H, J = 10.1 Hz, CH3O), 6.83 (d, 4 Harom., J = 8.5 Hz), 7.33 (dd, 4 Harom., J = 8.5/2.1 Hz)(AA´BB´-system). 13C NMR (75 MHz, CDCl3): d = 13.47 (s, 8 CH3(Bu)), 19.17 (s, 8 CH2(Bu)), 23.08 (s, 8 CH2(Bu)), 35.05 (d, J = 123.7 Hz, 2 CH2P), 50.47 (m, 2 CH3O), 57.42 (m, 8 CH2N), 128.39 (m, 4CHarom.), 134.50 (m, 2Carom.). 31P NMR (CDCl3): d = 19.34 (s). Elemental analysis: calcd. for C48H90N2O7P2 x 2H2O: C 63.69, H 10.47, N 3.09, found C 63.53, H 10.43, N 3.08.
1
H NMR Titrations.Procedure A: A solution of the phosphonate (10 equivalents in 0.4 mL of [D6]DMSO) was added with a 50 ml-microsyringe in aliquots to a solution of the alkyl guanidinium or amidinium chloride (1 equivalent in 0.7 mL of [D6]DMSO; 0.5-2 mM) in a sealed NMR-tube. The.guanidine solution contained ~ 0.05-0.1 % of water, the tetrabutylammonium phosphonate solution because of its highly hygroscopic character contained ~ 0.3-0.6 % of water. Volume and concentration changes were taken into account during analysis. NMR titrations on the 500 MHz spectrometer were carried out automatically by a roboter. To this end nine NMR-tubes were each filled with the same amount and concentration of guest solution, and increasing amounts of host solutions of uniform concentration were added successively. Errors in Ka were markedly smaller compared to the 300 MHz measurements due to the preparation of stock solutions and their tenfold lower concentrations.
Procedure B: Ten NMR tubes were filled each with 0.8 mL of a solution of the host compound (cguest = 0.5–4 mM) in a deuterated solvent (DMSO-d6,). The guest compound (1.525 equiv corresponding to the guest) is dissolved in 0.61 mL of the same solvent, and the resulting solution is added with increasing volumes from 0 to 5 equiv to the guest solution in ten NMR tubes.
Volume and concentration changes were taken into account during analysis. The association constants were calculated by non-linear regression methods.
1
H NMR Dilution Experiments.Two equivalents of 1 and 1 eqivalent of the respective amidine hydrochloride are dissolved in 1mL of DMSO-d6. 0.1 mL of this stock solution are then diluted to 1.4 mL (this produces a start concentration of 3 mM). For the first NMR measurement, 0.7 mL of this solution are used. The rest is then diluted again to 1.4 mL and the above described procedure is repeated 6 times, until a final concentration of 5 mM is reached. For the most diluted probe 5000 pulses are necessary (500 MHz spectrometer).
Supporting Information
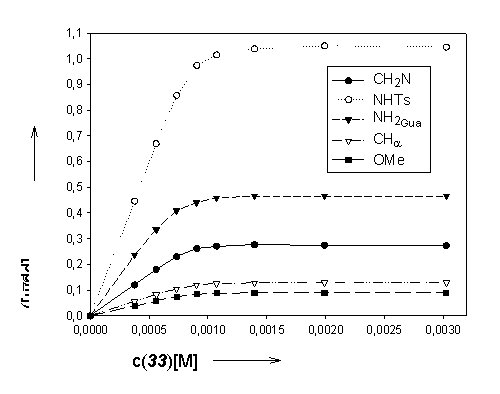
1. NMR-titration of N-a-tosylarginine methyl ester with m-xylylene bisphosphonate 1:
a) Host- / guest concentrations – chemically induced shifts (CIS) – binding constants
|
cG |
cH |
D CH2N |
D NHTs |
D NH2(Gua) |
D CHa |
D MeO |
K a / Error |
|
[M] |
[M] |
[ppm] |
[ppm] |
[ppm] |
[ppm] |
[ppm] |
[M-1] / % |
|
8,46e-4 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
86000 |
|
8,22e-4 |
3,78e-4 |
0,12 |
0,45 |
0,24 |
0,06 |
0,04 |
+/- 28% |
|
8,11e-4 |
5,60e-4 |
0,18 |
0,67 |
0,34 |
0,08 |
0,06 |
|
|
8,00e-4 |
7,36e-4 |
0,23 |
0,86 |
0,41 |
0,11 |
0,07 |
|
|
7,89e-4 |
9,07e-4 |
0,26 |
0,97 |
0,44 |
0,12 |
0,08 |
|
|
7,79e-4 |
1,08e-3 |
0,27 |
1,01 |
0,46 |
0,13 |
0,09 |
|
|
7,59e-4 |
1,40e-3 |
0,28 |
1,04 |
0,47 |
0,13 |
0,09 |
|
|
7,22e-4 |
1,99e-3 |
0,28 |
1,05 |
0,47 |
0,13 |
0,09 |
|
|
6,58e-4 |
3,03e-3 |
0,27 |
1,05 |
0,47 |
0,13 |
0,09 |
b) Graphical illustration: host concentration vs. CIS guest molecule
2. NMR titration of acetamidine hydrochloride with m-xylylene bisphosphonate 1:
a) Host- / guest concentrations – chemically induced shifts (CIS) – binding constants
|
cG |
cH |
D NH2i |
D NH2a |
D Me |
K a / Error |
|
[M] |
[M-1] |
[ppm] |
[ppm] |
[ppm] |
[M-1] / % |
|
1,53e-3 |
0,00 |
0,00 |
0,00 |
0,00 |
100.000 |
|
1,49e-3 |
7,43e-4 |
0,61 |
2,00e-3 |
0,15 |
+/-24 % |
|
1,47e-3 |
1,10e-3 |
0,78 |
0,03 |
0,22 |
|
|
1,45e-3 |
1,45e-3 |
0,87 |
0,07 |
0,28 |
|
|
1,43e-3 |
1,78e-3 |
0,89 |
0,11 |
0,31 |
|
|
1,41e-3 |
2,11e-3 |
0,90 |
0,12 |
0,32 |
|
|
1,37e-3 |
2,74e-3 |
0,90 |
0,13 |
0,33 |
|
|
1,31e-3 |
3,91e-3 |
0,90 |
0,14 |
0,33 |
|
|
1,19e-3 |
0,01 |
0,91 |
0,14 |
0,33 |
b) Graphical illustration: host concentration vs. CIS guest molecule

3. NMR titration of benzamidine hydrochloride with m-xylylene bisphosphonate 1:
a) Host- / guest concentrations – chemically induced shifts (CIS) – binding constants

b) Graphical illustration: host concentration vs. CIS guest molecule

4. NMR titration of acetamidine hydrochloride with diphenylether 2:
a) Host- / guest concentrations – chemically induced shifts (CIS) – binding constants

b) Graphical illustration: host concentration vs. CIS guest molecule
5. Dilution experiment of p-methoxybenzamidine hydrochloride with (1):
Amounts: 1.01 mg (5.41 µmol) p-Methoxybenzamidine hydrochloride 8.45 mg (10.87 µmol) (1)
The calculated association constant Ka (1 : 1) has a value of 76000 M-1 (+/- 13 %). Graphical representation of the titration curves:

6. Dilution experiment of benzamidine hydrochloride monohydrate with (1)
Amounts: 1.15 mg (6.585 µmol) benzamidine hydrochloride monohydrate 10.29 mg (13.171 µmol) (1)
The calculated association constant Ka (1 : 1) has a value of 120000 M-1 (+/- 17 %).
Graphical representation of the titration curves:
 |
 |
7. Dilution experiment of p-amidinobenzamide hydrochloride with (1)
Amounts: 1.13 mg (5.66 µmol) p-amidinobenzamide hydrochloride 8.88 mg (11.4 µmol) (1)
The calculated association constant Ka (1 : 1) has a value of 140000 M-1 (+/- 30 %).
Graphical representation of the titration curves:

8. Dilution experiment of m-nitrobenzamidine hydrochloride with (1)
Amounts: 0.93 mg (4.61 µmol) m-nitrobenzamidine hydrochloride 7.19 mg (9.25 µmol) (1)
The calculated association constant Ka (1 : 1) has a value of 250000 M-1 (+/- 20 %).
Graphical representation of the titration curves:

 |
 |
Table 1. Association constants (K
1:1) [M-1] from NMR titrations with 1 and 2 in DMSO at 20°C.[a]|
Guanidinium or amidinium derivative |
Receptor molecule |
(K 1:1)[M-1] |
D G [kcal/mol] |
|
N- a-tosylarginine methyl ester |
1 |
86000 ± 28% |
6.7 |
|
1,1-Dimethylguanidine |
1 |
36000 ± 38% |
6.2 |
|
Methylguanidine |
1 |
25000 ± 35% |
6.0 |
|
N- a-tosylarginine methyl ester |
2 |
21800 ± 13% |
5.8 |
|
1,1-Dimethylguanidine |
2 |
6600 ± 14% |
5.1 |
|
Methylguanidine |
2 |
5600 ± 39% |
5.0 |
|
Acetamidine (2:1-complex) |
2 |
1000 ± 20% |
4.0 |
a
Due to the strongly hygroscopic character of both titration partners the d6-DMSO solution contained ~0.1% of water. Errors in Ka are standard deviations and were calculated to be ± 6-39%.Table 2. Association constants (K
1:1) [M-1] from NMR-dilution experiments with 1 in DMSO at 20°C.[a]|
Amidine hydrochloride |
(K 1:1)[M-1] |
D G [kcal/mol] |
|
p -Methoxybenzamidine |
76000 ± 13% |
6.6 |
|
Acetamidine |
103000 ± 15% |
6.8 |
|
Benzamidine |
120000 ± 17% |
6.9 |
|
p -Amidinobenzamide |
140000 ± 30% |
7.0 |
|
m -Nitrobenzamidine |
250000 ± 20% |
7.3 |
a Due to the strongly hygroscopic character of both titration partners the d6-DMSO solution contained ~0.1% of water. Errors in Ka are standard deviations and were calculated to be ± 13-30%.
References
[1] Only the other way round is known: amidinium compounds as (chiral) hosts for anions and negatively polarized guests have been recently described: a) Y. Dobashi, . Kobayashi, N. Sato, A. Dobashi, Tetrahedron Lett. 1998, 39, 2985-2988; b) T. Schuster, M. W. Göbel, Synlett 1999, 966-968.
[2] T. Schrader, Chem. Eur. J. 1997, 3, 1537; T. Schrader, Tetrahedron Lett. 1998, 39, 517-520.
[3] Molecular Modeling Program: CERIUS2 from Molecular Simulations Inc., Force field: Dreiding 2.21.
[4] D. M. Perreault, L. A. Cabell, E. V. Anslyn, Bioorg. & Med. Chemistry 1997, 5, 1209-1220.
[5] a) A. W. Braithwaite, B. C. Baguley, Biochemistry 1980, 19, 1101-1106; b) M. L. Kopka, C. Yoon, D. Goodsell, P. Pjura, R. E. Dickerson, J. Mol. Biol. 1995, 183, 553-563 c) D. Goodsell, R. E. Dickerson, J. Med. Chem. 1986, 29, 727-733; d) M. Cory, R. R. Tidwell, T. A. Fairley, J. Med. Chem. 1992, 35, 431-438; e) B. Sellergren, Anal. Chem. 1994, 66, 1578-1582.
[6] J. W. H. M. Uiterwijk, C. J. van Staveren, D. N. Reinhoudt, H. J. den Hertog, Jr., L. Kruise, S. Harkema, J. Org. Chem. 1986, 51, 1574-1587.
[7] a) Schneider, H. J.; Kramer, R.; Simova, S.; Schneider, U. J. Am. Chem. Soc. 1988, 110, 6442. b) Macomber, R. S. J. Chem. Educ. 1992, 69, 375–378.
[8] J. P. Kirby, J. A. Roberts, D. G. Nocera, J. Am. Chem. Soc. 1997, 119, 9230-9236 and literature cited herein.
[9] b) Wilcox, C. S. in Frontiers in Supramolecular Chemistry and Photochemistry; Schneider, H.-J., Dürr, H., Eds.; VCH: Weinheim, 1991; pp. 123–143.
[10] T. W. Bell, V. J. Santora, J. Am. Chem. Soc. 1992, 114, 8300-8302.
[11] a) J. Shorter, in "Correlation Analysis in Chemistry", Plenum Press, New York, 1978; b) M. Charton, Prog. Phys. Org. Chem. 1981, 13, 119; c) W. F. Reynolds, Prog. Phys. Org. Chem. 1983,14, 165.
[12] U. Obst, Dissertation Nr. 12037, ETH Zürich, Switzerland.
[13] K. J. Edwards, T. C. Jenkins, S. Neidle, Biochemistry 1992, 31, 7104-7109.
[14] a) D. G. Brown, M. R. Sanderson, E. Garman, S. Neidle, J. Mol. Biol. 1992, 226, 481-490; b) C. A. Laughton, F. Tanious, C. M. Nunn, D. W. Boykin, W. D. Wildon, S. Neidle, Biochemistry 1996, 35, 5655-5661.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with C0027 as the message subject of your e-mail.
