
[C0034]
A MODEL STUDY FOR A NOVEL SYNTHETIC APPROACH TO 2-CARBOXYINOSINES
Vasanthakumar Rajappan and Ramachandra S. Hosmane*
Laboratory for Drug Design and Synthesis
Department of Chemistry and Biochemistry
University of Maryland, Baltimore County, 1000 Hilltop
Circle
Baltimore, Maryland 21250
* Phone: 410-455-2520 Fax: 410-455-1148 E-Mail:
[email protected]
Received: 14 August 2000 / Uploaded: 16 August
Key Words: hypoxanthines, 2-substitution, 2,2-disubstitution, 2-carboxyhypoxanthine, 2,2-dicarboxylic acid ester
ABSTRACT
A model study for a novel approach to the synthesis
of 2-carboxyinosines has been described. The synthesis of 9-benzyl-2-carboxyhypoxanthine
(I), a model for 2-carboxyinosine, was achieved in 4 steps starting
from 1-benzyl-5-nitroimidazole-4-carboxylic acid. Also reported is the
synthesis of 9-benzyl-2,2-bis(ethoxycarbonyl)hypoxanthine (II).
INTRODUCTION
There has been continued biomedical interest in the synthesis
of analogues of inosine substituted at the 2-position with a functional
group that is a member of the carboxylic acid family, including the parent
carboxylic acid moiety,1 a carboxamide group,2 a
nitrile,1,3 an imidate,1 or an ester functionality.4
Also reported are inosines or 2'- or 3'-deoxyinosines or hypoxanthines
substituted with a carboxaldehyde or a ketone group at the 2-position.3,5
Most of these reported methods involve direct functional group transformations
of nucleosides. While the use of nucleosides has an advantage in that it
forgoes the glycosylation step, it nevertheless suffers from extensive
protection and deprotection steps of not only the heterocyclic moiety,
but also of the sugar hydroxyls. This is especially true in case of 2-substituted
purines since the formation of a C-C bond by direct nucleophilic displacement
reaction, for example, of a 2-halo substituent by cyanide anion, has largely
been unsuccessful.1,2 This often necessitates organometallic
reactions that call for stringent requirements of protection/deprotection
steps, such as the Stille coupling6 between an organic iodide
and tri-n-butyl cyanide in the presence of tetrakis(triphenylphosphine)
palladium. Thus the reported conversion of guanosine into 2-cyano-3'-deoxyinosine,
an analogue of the naturally occurring antibiotic cordycepin,7
involved ten synthetic steps.3 Although sulfonylmethyl1
and sulfonylphenyl2 groups at the purine 2-position have been
reported to be far better leaving groups than the corresponding halogen
substituents, the attempted direct displacement reactions by a cyanide
or other carbanions on the sulfonyl substituents failed on both guanosine1
and inosine.2 The explanation given for these failures is that
the dissociable N-1 proton in either guanosine or inosine, especially at
a reasonably high temperature that is normally employed for such displacement
reactions (~100 C in DMF), renders the C-2 position less susceptible for
nucleophilic attack.1,2 The support for such a notion came from
the successful nucleophilic displacement reactions with 2-sulfonylalkyl
or 2-sulfonylaryl derivatives of either adenosine1 or 6-methoxypurine.2
We report here an alternative short approach to the synthesis of 2-substituted hypoxanthines. The synthesis of 9-benzyl-2-carboxyhypoxanthine (I), described herein, is a heterocyclic model for 2-carboxyinosine. Since the parent carboxylic acid group can be conveniently converted to other members of the carboxylic acid family as well as to aldehydes and ketones employing conventional chemical methods, the synthesis is potentially versatile. Also reported here is the synthesis of 2,2-diester-substituted analogue II that was obtained by mere change of solvent in the final ring-closure step. Although the initial purpose of its synthesis was to confirm the anticipated mechanism of formation of I, compound II is apparently a good precursor to a wide variety of 2,2-disubstituted analogues of hypoxanthine and inosine, which may otherwise be difficult to prepare by usual synthetic methods.

The synthesis of target I was achieved in four steps commencing from 1-benzyl-5-nitroimidazole-4-carboxylic acid (1)8 (Scheme I). Condensation of 1 with diethylaminomalonate, mediated by 1,1'-carbonyldiimidazole (CDI), yielded diethyl 2-[N-(1-benzyl-5-nitroimidazolyl-4-carbonyl)amino]malonate (2) in 93% yield, mp 88 C; 1H NMR (300 MHz, DMSO-d6) 9.27-9.29 (d, J = 7.2 Hz, 1H, NH, ex./ w. D2O), 8.28 (s, 1H, CH), 7.10-7.83 (m, 5H, Ar-H), 5.54 (s, 2H, NCH2 ), 5.22-5.24 (d, JNH-CH = 7.2 Hz, 1H, CH ), 4.18 (q, 4H, 2xCH2), 1.20 (t, 6H, 3xCH3); Anal. C, H, N.9 The reaction of 2 with bromine in the presence of sodium hydride in dimethylformamide, followed by treatment with potassium phthalimide afforded diethyl 2-phthalimido-2-[N- (1-benzyl-5-nitroimidazolyl-4-carbonyl)amino]malonate (3) in 92% yield, mp 83-85 oC; 1H NMR (300 MHz, DMSO-d6) d 9.06 (s, 1H, NH), 8.26 (s, 1H, CH), 7.92 (m, 4H, Ar-H), 7.35 (m, 5H, Ar-H), 5.48 (d, 2H, NCH2), 4.30 (q, 4H, 2XCH2), 1.10 (t, 6H, 2XCH3); Anal. C, H, N.9 This reaction is presumed to proceed (Scheme II) via base-catalyzed bromination of the side-chain malonate moiety to produce 6, followed by dehydrohalogenation to form the

imine intermediate 7. The latter would readily react with the phthalimide anion to produce 3. The nitro group of 3 was reduced by catalytic transfer hydrogenation10 using cyclohexene and palladium on charcoal, which gave diethyl 2-phthalimido-2-[N-(5-amino-1-benzylimidazolyl-4-carbonyl)amino] malonate (4) in 45% yield, mp 162 - 164 oC; 1H NMR (300 MHz, DMSO-d6) d 9.22 (s, 1H, NH), 8.19 (s, 1H, CH), 7.59-7.18 (m, 9H, phenyl H), 5.94 (s, 2H, NH2), 5.10 (s, 2H, NCH2), 4.22 (q, 4H, 2XCH2), 1.15 (t, 6H, 2XCH3); Anal. C, H, N.9 Finally, the target 9-benzyl-2-carboxyhypoxanthine (I) was prepared in 78% yield by heating 4 at 70 C for ten minutes in anhydrous dimethyl sulfoxide containing slightly over two equivalents of sodium hydride, mp >200 C; 1H NMR (300 MHz, DMSO-d6) d 13.56 (br, s, 1H, CO2H), 8.43 (s, 1H, NH), 8.12 (s, 1H, CH), 7.38-7.21 (m, 5H, Ar-H), 5.05 (s, 2H, CH2); Anal. C, H, N.9
The conversion of 4 into I is believed to go through an imine intermediate 8 (Scheme III), formed by base-catalyzedelimination of phthalimide, followed by ring-closure to produce II in situ. The latter undergoes facile nucleophilic
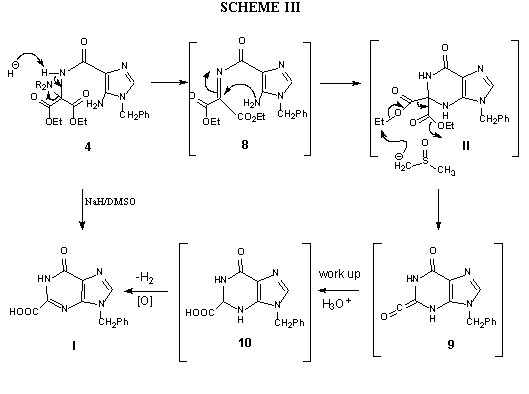
displacement by the dimsyl anion present in the medium
with concomitant elimination of carbon dioxide to form a ketene-type intermediate
9. The aqueous work-up of 9 to produce 10, accompanied
by spontaneous aromatization of the latter by loss of molecular hydrogen,
yields the target 2-carboxyinosine analogue I. The support for the
speculated mechanism came from the ring-closure reaction of 4 carried
out in in a solvent that is non-nucleophilic or is incapable of forming
a nucleophilic species by reaction with with sodium hydride, such as the
dimsyl anion mentioned above. Accordingly, when the final ring-closure
of 4 was carried out in dimethylformamide instead of dimethyl sulfoxide
as the solvent, it gave 9-Benzyl-2,2-bis(ethoxycarbonyl)hypoxanthine (II),
mp 166-171 oC; 1H NMR (300 MHz, DMSO-d6)
10.06 (s, 1H, NH), 9.57 (s, 1H, NH), 8.23 (s, 1H, CH), 7.42 (m, 5H, Ar-H),
5.18 (s, 2H, CH2), 4.18 (q, 4H, 2xCH2), 1.62 (t,
6H, 2xCH3); Anal. C, H, N.9 The speculated intermediacy
of II in the conversion of 4 to I was further corroborated
by a separate treatment of the isolated II with sodium hydride in
dimethyl sulfoxide under identical reaction conditions as used for 4
I conversion, which indeed afforded I.
ACKNOWLEDGMENT
This research was supported by a grant from the National
Institutes of Health (# CA 71079). The mass spectra were run at the MSU-NIH
Mass Spectral Facility, supported by an NIH grant (#P41 RR00480).
REFERENCES AND NOTES
1. Matsuda, A.; Nomoto, Y.; Ueda, T. Chem. Pharm. Bull. 1979, 27, 183.
2. Matsuda, A.; Satoh, K.; Miyasaka,
T.; Ueda, T. Chem. Pharm. Bull. 1984,
32,
2048.
3. Nair, V.; Lyons, A. Tetrahedron 1990, 46, 7677.
4. Bhan, A.; Hosmane, R. S. J. Heterocyclic Chem. 1993, 30, 1453.
5. Kehler, J.; Henriksen, U.; Vejbjerg,
H.; Dahl, O.; Bioorg. Med. Chem.
1998,
6, 315.
6. Stille, J. K. Angew. Chem. Int. Ed. Engl. 1986, 25, 508.
7. Suhadolnik, R. J., Ed. Nucleosides
as Biological Probes, Wiley
Interscience:
New York, 1979; p. 118.
8. Hosmane, R. S.; Bhan, A. J. Heterocyclic Chem. 1990, 27, 2189.
9. The elemental microanalyses were
performed by Atlantic Microlab, Inc.,
Norcross,
Georgia, and the analyses were found to be within 0.4% of the
calculated
values.
10. Bianco, A.; Passacantilli, P.; Righi, G. Tetrahedron Lett. 1989, 30, 1405.
All comments on this poster should be sent by e-mail to (mailto:[email protected] ona.edu)
[email protected] with C0034 as the message subject of your e-mail.