e-mail: [email protected]
![]()
Thienylsubstituted Derivatives of a-Aminobutanoic
acid.Practical
Approach to Enantiomerically Pure g-Hydroxy-a-aminooctanoic
and g-Hydroxy-a-aminononanoic
Acids.
e-mail: [email protected]
![]()
The initial g-thienylsubstituted-g-oxo-a-amino acids (2a-c) were prepared by the conjugate addition of benzylamine or 1-phenylethylamine to appropriate thenoylacrylic acid (1A,B). This reaction is regioselective (only derivatives of a-amino acids are formed) and using a chiral nonracemic amines also highly enantioselective.6a,b The thenoylacrylic acids (1A,B) are accessible by most common and frequent method - Friedel-Crafts reaction of maleic anhydride and relevant thiophene derivative in the presence of AlCl3 (Scheme 1).
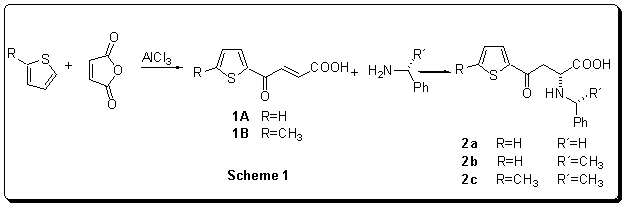
Table
1Results
of Friedel-Crafts reaction and the conjugate addition of benzylamine or
1-phenylethylamine
|
|
|
|
(%) |
|
(°C) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The first method of the preparation of
enantiomerically pure thienylsubstituted syn-g-hydroxy-a-aminobutanoic acids, using
L-kynurenine hydrolase was described in 1998.7 The diastereomeric
ratio of the products was 95:5 for syn-g-hydroxy-a-aminobutanoic acid isolated in 46 %
yield.
Recently we have published8 the
stereoselective catalytic reduction of the g-aryl-g-oxo-a-amino acids that allowed us to
reach a d.r. of up to 97:3 with syn-g-hydroxy-a-amino acids as the major products.
According to this method the syn-g-thienylsubstitutedg-hydroxy-a-amino acids (3a-c) were
prepared in a high yield and diastereoselectivity (Scheme 2). In comparison with
up to present known methods this reduction is marked out with its simplicity of
implementation and high stereoselectivity. Other methods, using complex hydrides
for the reduction of carbonyl group allow to obtain products with much lower
diastereoselectivity 9-12. Esters and amides of g-oxo-a-amino acids under these conditions
give a mixture of both diastereomers most frequently in the form of relevant
lactones. 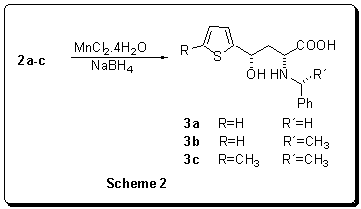
Table
2Results
of the stereoselective sodium borohydride reduction, catalyzed by manganesse(II)
chloride
|
|
|
|
(%) |
|
(°C) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hardly any attention was paid to the
lactonization of free g-hydroxy-g-aryl-a-amino acids. The group of Prof.
Jäger in his work presents a strong dependence of the diastereomeric ratio
(cis- and trans- lactones) on reaction conditions.13
Recently we have presented the method of stereoconvergent lactonization
of g-aryl substituted, g-hydroxy-a-aminobutanoic acids represents an
another example of CIDR.14Application of this method into the
thienyl substituted derivatives allowed us to prepare adequate
cis-g-thienyl-a-aminobutanolides (4a-c). Its
alkaline hydrolysis opened up the access to the relevant
anti-g-thienyl-g-hydroxy-a-aminobutanoic acids
(5b,c)(Scheme 3).
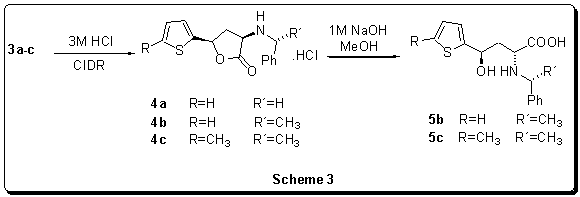
Table
3Results
of the stereoconvergent lactonization
|
|
|
|
(%) |
|
(°C) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Table
4anti-g-Thienylsubstituted-a-phenylethylaminobutanoic
acids prepared by alkaline hydrolysis
|
|
|
|
(%) |
|
(°C) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Forasmuch as the thiophene ring in the
prepared g-thienylsubstituted-g-hydroxy-a-phenylethylaminobutanoic acids
represents a precursor of C-4 aliphatic chain, its reduction using Raney
nickel could be the pivotal step of the synthesis of enantiomerically pure
aliphatic derivatives of amino acids.
The desulfurization of the thiophene ring has
been studied by several research groups15-18. Application of
Raney nickel allowed them to obtain an aliphatic part of carbon chain.
The only known work describing the desulfurization of hydroxyaminocarboxylic
acid uses a great excess of Raney nickel for desulfurization of racemic
thienyl serine and the reaction run with a low yield
19.
Enantiomerically pure derivatives
of a-aminooctanoic, or a-aminononanoic acids are a useful
building blocks of many biologically potent derivatives20-22.
Moreover the lactones of higher g-hydroxy-a-aminocarboxylic acids are
substructural units of substances showing a strong ACE inhibiting
effect.23 The only known method for the preparation of these lactones
in the racemic form is the isoxazoline route24.
Our improved method of desulfurization g-thienylsubstituted-g-hydroxy-a-aminobutanoic acids (4a-c; 5b,c) was carried out in methanol in the hydrogen atmosphere and with a gradual addition of Raney nickel. The best results (yield, diastereomeric purity, time demands) was achieved within the series of syn-derivatives. This way prepared aliphatic syn-g-hydroxy-a-phenylethylaminooctanoic, or aminononanoic acids (6b,c) and its anti-diastereomers (7b,c) were put through debenzylation under the catalytic influence of Pd(OH)2/C in the hydrogen atmosphere. Relevant syn-, or anti-g-hydroxy-a-aminooctanoic, or aminononanoic acids (8b,c; 9b,c)were isolated in a high yield (Scheme 4).
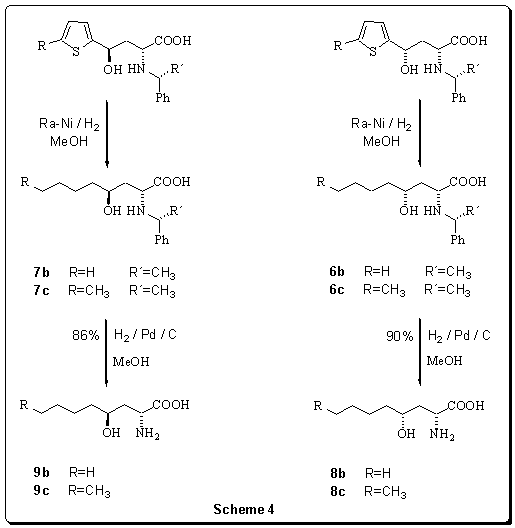
Table
4Results
of the desulfurization of the thiophene ring using Raney
nickel.
|
|
|
|
(%) |
|
(°C) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The structure of synthesized compounds was confirmed by 1H NMR and 13C NMR spectroscopy and basis of appointed physico-chemical constants.
Experimental
General
methods
All
reagents were used as received without further purification unless otherwise
specified. Melting points were obtained using a Büchi 510 capillary melting
point apparatus and a Kofler hot plate and are uncorrected. Optical rotations
were measured with a Perkin-Elmer 241 polarimeter and a POLAR L-mP
polarimeter (IBZ Messtechnik) with a water-jacketed 10.000 cm cell at a
wavelength of sodium line D (l
= 589 nm). Specific rotations are given in units of 10-1
deg.cm2.g-1 and concentrations are given
in g/100 ml. 1H NMR spectra were recorded on a Varian VXR-300 (299.94
MHz) spectrometer. Chemical shifts (d)
are quoted in ppm and are either referenced to the tetramethylsilane (TMS) as
internal standard (dMe =
0.00 ppm for 299.94 MHz) or the residual protic solvent
(CH3OH, dH =
3.34 ppm for 299.94 MHz) was used as the internal reference. Coupling constants
(J) are recorded in Hz. The following abbreviations were used to
characterize signals muliplicities : s (singlet), d (doublet), t (triplet), m
(multiplet), b (broad). Abbreviations with quotation marks mean that the
appearance of the signal is different of that theoretically predicted.
13C NMR spectra were recorded on a Varian VXR-300 (75.43 MHz)
spectrometer. The multiplicities of carbons were assigned from a broadband
decoupled analysis used in a conjunction with either APT or DEPT programs.
Chemical shifts are quoted in ppm and are either referenced to the
tetramethylsilane (TMS) as internal standard (d=
0.00 ppm for 75.43 MHz) or the central resonance of CD3OD
(d=
49.0 ppm for 75.43 MHz) were used as the internal reference. The following
abbreviations were used to characterize signals muliplicities : s (singlet), d
(doublet), t (triplet), q (quartet).
b-Heteroaroylacrylic
acids (1A, B)
To
a stirred suspension of dichloromethane (200ml) and anhydrous AlCl3
(53.2g, 0.4mol) gradually maleic anhydride (15.7g, 0.16mol) was added. The
temperature could not exceed 25-30°C and the mixture was stirred at 25°C for 30
min.
Then
the mixture was cooled to cca 15°C and a corresponding thiophene (0,16mol) in
dichlormethane (100ml) was added dropwise to a reaction mixture (20 min). After
the addition the temperature was maintained at 15°C for another 1 hour. The
decomposition of the reaction mixture was achieved with crushed ice (300g) and
concentrated hydrochloric acid (30ml). Subsequent extraction with dichlormethane
(3´100ml),
drying over anhydrous sodium sulfate and the evaporating of the solvent afforded
to white solid. The obtained crude product was crystallized from
toluene.
4-(2-thienyl)-4-oxo-2-butenoic
acid (1A)
m.p.
152-153.5°C
13C
NMR (CDCl3/CD3OD): 181.6 (C-6); 167.4 (C-1); 144.3
(C-2)
1H
NMR (CDCl3/CD3OD): 7.89 (d, J=3.9 Hz, J=1.0
Hz, H-5´´); 7.80 (d, 15.5 Hz, H-2); 7.79 (dd, J=5.0 Hz,J=1.0 Hz,
H-3´´); 7.21 (dd, J=5.0 Hz,J=3.9 Hz, H-4´´); 6.92 (d,
J=15.5 Hz, H-3);
4-(2-(5-methylthienyl)-4-oxo
2-butenoic acid (1B)
m.p.
150-155°C
1H
NMR (CDCl3/CD3OD): 7.98 (d, J=4.9 Hz, H-3´´); 7.73
(d, J=19.4 Hz, H-3); 6.97 (d, J=4.9 Hz, H-4´´); 6.63 (d,
J=19.4 Hz, H-2); 2.5 (s, 3H, CH3)
g-Oxo-a-amino
acids (2a-c)
A
relevant b-heteroaroylacrylic
acid (0.02mol) was dissolved in MeOH (50 ml) and benzylamine (2.2g, 0.02mol), or
(R)-1-phenylethylamine (2.4g, 0.02mol) with triethylamine (2g, 0.02mol) was
added. The mixture was stirred at r.t. for 48 72 hours and the process was
monitored by HPLC. The precipitate was filtered off, washed with methanol
(2´10ml),
ether (10ml) and dried to afford 4.33 g (75%) of 2a, 5.57 g (92%) of
2b, or 5.18 (80%) of 2c as a white solid.
4-(2-thienyl)-4-oxo-2-(benzylamino)butanoic
acid (2a)
m.p.
185-186°C
13C
NMR (NaOD/D2O): 197.4 (C-4); 182.8 (C-1); 145.7 (C-2´´); 141.6
(C-1-Ph); 138.8 (C-5´´);
137.5 (C-3´´);
131.8 (C-4´´);
131.4,131.3 (C-2,3,5,6-Ph); 130.1 (C-4-Ph); 62.5 (C-2); ~45.0 (C-3);
1H
NMR (NaOD/D2O):
7.75-7.95 (m, 2H, H-5´´, H-3´´);
7.2-7.5 (m, 5H, Haromat.);
7.19 (bs, 1H, H-4´´);
3.75 (d, J=12.9 Hz, 1H, H-1´);
3.59 (bs, 1H, H-2);
3.3-3.1 (m, 0.2 H, H-3)
(2R,1´R)-4-(2-thienyl)-4-oxo-2-(1´-phenylethylamino)butanoic
acid (2b)
m.p.
194-196°C
13C
NMR (CD3OD/DCl): 189.7 (C-4); 170.5 (C-1); 143.0 (C-2´´); 136.8
(C-5´´); 136.6 (C-1-Ph); 135.3 (C-3´´); 131.0 (C-4´´); 130.6,129.2
(C-2,3,5,6-Ph); 129.8 (C-4-Ph); 60.0 (C-1´); 54.5 (C-2); 40.0 (C-3); 20.4
(C-2´)
1H
NMR (CD3OD/DCl): 7.94 (d, J=4.5 Hz, 2H, H-3´´, 5´´); 7.42-7.65
(m, 5H, Harom.); 7.25 (dd, J=4.5 Hz, 1H, H-4´´); 4.74 (q,
J=6.9 Hz, 1H, H-1´);4.12 (t,
J=5.1 Hz, 1H, H-2); 3.62-3.84 (m, 2H, H-3); 1.79 (d, J=6.6 Hz, 3H,
H-2´)
13C
NMR (DCl/D2O): 132. 0, 130.4 (C-2,3,5,6-Ph);
66.7 (C-1´, C-2);
61.7 (C-1´, C-2);
40.4 (C-3);
21.6 (C-2´´);
17.9 (CH3-Tio)
1H NMR (DCl/D2O): 7.15 (bs, 1H,
H-3´´); 7.1-6.92 (m, 5H,
Harom.); 6.42 (bs, 1H, H-4´´); 4.19 (q, J=6.6 Hz, 1H,
H-1´); 3.71 (m, 1H, H-2); 3.29-3.00 (m, 1H, H-3); 2.02 (s,3H,
CH3-Tio); 1.26 (d, J=6.9 Hz, 1H,
H-2´
syn-g-Hydroxy-a-amino
acids(3a-c)
To
a presonicated (1 min) stirred suspension of relevant g-oxo-a-amino
acid 2a-c (10 mmol) and MnCl2.4H2O (0.4g, 2mmol) in
MeOH (15ml) at 5°C NaBH4 (2´0.37g,
2´10mmol)
was slowly added (during 2´10
min)(Note 1.). The resulting solution was then stirred for 30 min and
H2O (100ml) was added. Methanol was removed under reduced pressure
and the pH of the remaining solution was decreased to 6.0 with 1N HCl. A
precipitate was filtered off, washed with water (20ml), MeOH (10ml), ether (5ml)
and dried. The crude product was crystallized from 70% EtOH.(Note
2.)
Note
1.: In case of 3b, 3c it was needful for the full conversion of the
reaction more than 2 equivalents of NaBH4.
Note
2.: 3b generates in organic solvents stable gel, data in
experimental are taken into consideration as the crude product after
drying.
(2R*,4R*)-4-(2-thienyl)-4-hydroxy-2-(benzylamino)butanoic
acid (3a)
Yield
2.02 g (69%), m.p. 198-199°C
13C
NMR (NaOD/D2O): 184.2 (C-1); 150.0 (C-2´´); 141.6 (C-1-Ph); 131.6,
131.5 (C-2,3,5,6-Ph); 130.2 (C-4-Ph); 129.8 (C-5´´); 128.0, 127.4 (C-3´´,4´´);
70.8 (C-4); 63.6 (C-2); 54.0 (C-5); 44.6 (C-3)
1H
NMR (NaOD/D2O): 7.30-7.47 (m, 6H, Harom.,H-5´´); 7.00 (dd,
J=3.6 Hz, J=5.1Hz, 1H, H-4´´); 6.94 (bd, J=3.3 Hz, 1H,
H-3´´); 5.06 (t, J=6.9 Hz, 1H, H-4); 3.79 (q, J=12.7 Hz,
J=12.7 Hz, 1H, H-1´); 3.53 (q, J=12.7 Hz, J=12.7 Hz, 1H,
H-1´); 3.12 (t, J=6.9 Hz,1H, H-2); 2.00-2.10 (m, 2H,
H-3).
(2R,4S,1´R)-4-(2-thienyl)-4-hydroxy-2-(1´-phenylethylamino)butanoic
acid (3b)
Yield
2.27 g (73%), m.p. 199-200°C
[a ]D26 = +35.2 (c=1.1, 0.1 N
NaOH/H2O)
13C
NMR (NaOD/D2O): 184.4 (C-1); 149.5 (C-2´´); 146.6 (C-1-Ph);
130.1-131.5 (C-2,3,5,6-Ph); 130.3 (C-4-Ph); 129.6 (C-5´´); 128.0 (C-4´´); 127.5
(C-3´´); 71.2 (C-4); 62.7 (C-2); 59.4 (C-1´); 44.6 (C-3); 26.1
(C-2)
1H
NMR (NaOD/D2O): 7.30-7.47 (m, 6H, Harom.,H-5´); 6.95 (dd,
J=3.3 Hz, J=5.4Hz, 1H, H-4´´); 6.80 (dd, J=3.3 Hz,
J=1.2 Hz, 1H, H-3´´); 4.96 (dd, J=7.2 Hz, J=6.7, 1H, H-4);
3.69 (q, J=6.6 Hz, 1H, H-1´); 2.84 (dd, J=8.2 Hz, J=5.6 Hz,
1H, H-2); 1.85-2.05 (m, 2H, H-3); 1.39 (d, J=6.6 Hz, 3H,
H-2)
(2R,4S,1´R)-4-(2-(5-methyl-2-thienyl))-4-hydroxy-2-(1´-phenylethylamino)butanoic
acid (3c)
Yield
2.77 g (86%), m.p. 192-193°C
[a ]D26 = +38.1 (c=0.9,
MeOH)
13C
NMR (NaOD/D2O): 184.5 (C-1); 146.9 (C-Tio); 146.6 (C-1-Ph); 143.1
(C-Tio); 131.6 (C-2,6-Ph); 130.4 (C-4-Ph); 130.2 (C-3,5-Ph); 127.7-127.5
(C-Tio); 71.3 (C-4); 62.7 (C-2); 59.5 (C-1´); 44.5 (C-3); 26.1 (C-2);17.2
(C-Tio)
1H NMR (NaOD/D2O): 7.28-7.48
(m, 5H, Harom.); 6.59 (bs, 1H, H-4´´); 6.55 (bd, J=3.0 Hz, 1H,
H-3´´); 4.85 (t, 1H, H-4); 3.68 (q, J=6.6 Hz, 1H, H-1´); 2.81 (dd,
J=8.1 Hz, J=5.7 Hz, 1H, H-2); 2.41 (s, 3H, H-CH3-Tio);
1.75-2.00 (m, 2H, H-3); 1.38 (d, J=6.6 Hz, 3H,
H-2)
a-Aminobutanolides
(4a-c)
To
the relevant g-hydroxy-a-amino
acid 3a-c (4mmol) suspended in water (5ml) 4N HCl (30ml) at one portion
was added and the reaction mixture was stirred at room temperature. After
requested time (5-12 hours, HPLC control), the precipitate was filtered off,
dried and crystallized from appropriate solvent.
(2R*,4R*)-4-(2-thienyl)-2-(benzylamino)-4-butanolid
hydrogenchloride (4a)
Yield
0.91g (77%), m.p. 186-188°C
13C
NMR (DMSO-d6): 171.0 (C-1); 139.6 (C-2´´); 131.6 (C-1-Ph);130.3
(C-3,5-Ph);
129.2 (C-4-Ph); 128.8 (C-2,6-Ph); 128.4 (C-5´´); 128.2 (C-Tio); 127.3 (C-Tio);
74.5 (C-4); 54.5 (C-2); 48.7 (C-1´); 34.5 (C-3)
1H
NMR (DMSO-d6): 7.69 (d, J=I5.1 Hz, 1H, H-5´´); 7.67-7.59 (m,
2H, Haromat.);7.50-7.42 (m, 3H, Harom.); 7.11 (dd,
J=3.6 Hz, J=5.1 Hz, 1H, H-4´´); 5.85 (dd, J=5.5 Hz, 1H,
H-4); 4.59 (dd, J=8.5 Hz, J=3.4 Hz, 1H, H-2); 7.36 (dd,
J=13.0 Hz, J=16.6 Hz, 1H, H-1´); 3.18-3.00 (m, 1H, H-3); 2.85-2.65
(m, J=5.4 Hz, J=8.3 Hz, 1H, H-3)
(2R,4R,1´R)-4-(2-thienyl)-2-(1´-phenylethylamino)-4-butanolid
hydrogenchloride (4b)
Yield
0.95g (73%), m.p. 187-191°C
[a ]D26 = 31.3 (c=0.9,
MeOH)
13C
NMR (DMSO-d6): 171.0 (C-1); 139.6 (C-2´´); 136.6 (C-1-Ph);129.2
(C-4-Ph);
129.0 (C-3,5); 128.3 (C-5´´); 128.2 (C-2,6-Ph); 128.1 (C-3´´); 127.3 (C-4´´);
74.5 (C-4); 56.0 (C-2); 52.9 (C-1´); 35.0 (C-3); 19.3 (C-2)
1H
NMR (DMSO-d6): 10.3 (bs, 2H, NH2+);7.68 (bd,
1H, J=5.0 Hz, H-5´´);7.60-7.67 (m, 2H, Harom.); 7.40-7.56 (m,
3H, Harom.); 7.31 (bd, J=3.0 Hz, 1H, H-3´´); 7.09 (dd, 1H,
J=4.8 Hz, J=3.9 Hz, H-4´´); 5.76 (dd, J=5.4 Hz,
J=10.8 Hz, 1H, H-1´); 4.37 (m, 1H, H-2); 2.88-2.57 (m, 1H, H-3); 1.64 (d,
J=6.9 Hz, 3H, H-2)
(2R,4R,1´R)-4-(5-methyl-2-thienyl))-2-(1´-phenylethylamino)-butanolid
hydrochloride (4c)
Yield
0.96g (71%), m.p. 169-173°C
[a ]D26 = 45.8 (c=1,
MeOH)
13C
NMR (CDCl3/CD3OD): 170.2 (C-1); 142.7 (C-2´´); 135.5
(C-5´´); 135.1 (C-1-Ph); 129.9 (C-4-Ph); 129.6-128.6 (C-2,3,5,6-Ph); 128.3
(C-3´´); 125.3 (C-4´´); 75.5 (C-4); 57.5 (C-1´); 52.8 (C-2); 35.2 (C-3); 20.5
(C-2); 15.4 (C-CH3-Tio)
1H
NMR (CDCl3/CD3OD): 7.70-7.75 (m, 2H, Harom.);
7.40-7.52 (m, 3H, Harom.); 6.95 (d, J=3.6 Hz, 1H, H-3´´); 6.57
(dq, 1H, J=3.6 Hz, J=1.2 Hz, H-4´´); 5.39 (dd, J=5.7 Hz,
J=10.2 Hz, 1H, H-4); 5.24 (bq, 1H, J=6.6 Hz, H-1´); 3.77 (dd, 1H,
J=8.4 Hz, J=12.0 Hz, H-2); 2.9-3.1 (m, 2H, H-3); 2.94 (s, 3H,
H-CH3-Tio); 1.88 (d, 3H, J=6.8 Hz,
H-R´)
anti-g-Hydroxy-a-aminobutanoic
acids (5b,c)
Relevant a-aminobutanolide
4b,c (2mmol) was dissolved in MeOH (25 ml) - H2O (15ml)
mixture and 1N NaOH (4ml) was slowly added under stirring. The reaction mixture
was stirred additionally 1 hour, MeOH was removed under reduced pressure and the
pH of the remaining water suspension was adjusted to 6.0 with 1N HCl. The
precipitated crude product was filtered off and dried under reduced
pressure.
(2R,4R,1´R)-4-(2-thienyl)-4-hydroxy-2-(1´-phenylethylamino)butanoic
acid (5b)
Yield
0.51g (82%), m.p. 192-194°C
[a ]D26 = +23.0 (c=1, 0.1 N
NaOH/H2O)
13C
NMR (NaOD/D2O): 185.4 (C-1); 150.7 (C-2´´); 146.6 (C-1-Ph);
129.9-131.4 (C-2,3,5,6-Ph); 130.1 (C-4-Ph); 129.6 (C-5´´); 127.6 (C-4´´); 127.2
(C-3´´); 70.2 (C-4); 61.7 (C-2); 59.1 (C-1´); 45.0 (C-3); 26.2
(C-2´)
1H
NMR (NaOD/D2O): 7.20-7.40 (m, 6H, Harom.,H-5´´); 6.92 (dd,
J=3.6 Hz, J=5.1Hz, 1H, H-4´´); 6.86 (dd, J=3.6 Hz,
J=1.2 Hz, 1H, H-3´´); 4.91 (t, 1H, H-4); 3.68 (q, J=6.6 Hz, 1H,
H-1´); 2.82 (t, J=6.8 Hz, 1H, H-2); 1.93-2.12 (m, 2H, H-3); 1.33 (d,
J=6.6 Hz, 3H, H-2´)
(2R,4R,1´R)-4-(5-methyl-2-thienyl)-4-hydroxy-2-(1´-phenylethylamino)butanoic
acid (5c)
Yield 0.45g (70%), m.p.
193-194°C.
[a ]D26 =+21.2 (c=1, 0.1 N
NaOH/H2O)
13C
NMR (NaOD/D2O): 184.5 (C-1); 147.2 (C-Tio); 146.6 (C-1-Ph); 142.9
(C-Tio); 131.5 (C-2,6-Ph); 130.2 (C-4-Ph); 130.1 (C-3,5-Ph); 127.7-127.5
(C-Tio); 70.4 (C-4); 61.7 (C-2); 59.3 (C-1´); 44.5 (C-3); 26.2 (C-2´);17.2
(C-Tio)
1H NMR (NaOD/D2O):7.25-7.45 (m, 5H, Harom.); 6.67 (bd,
J=2.5 Hz, 1H, H-3´´); 6.60 (bs, 1H, H-4´´); 4.85 (t, 1H, H-4); 3.70 (q,
J=6.8 Hz, 1H, H-1´); 2.82 (t, J=6.6 Hz, 1H, H-2); 2.42 (s, 3H,
H-CH3-Tio); 1.93-2.12 (m, 2H, H-3); 1.36 (d, J=6.5 Hz, 3H,
H-2´)
(2R,4R,1´R)-4-hydroxy-2-(1´-phenylethylamino)octanoic
acid (6b)
13C
NMR (NaOD/D2O): 184.9 (C-1); 146.8 (C-1-Ph); 131.6 (C-3,5-Ph); 130.4
(C-4-Ph); 130.2 (C-2,6-Ph); 74.1 (C-4); 63.5 (C-1´); 59.4 (C-2); 42.4 (C-3);
38.7 (C-5);29.7 (C-6); 26.3 (C-2´); 24.8 (C-7); 16.1 (C-8)
1H
NMR (NaOD/D2O):
7.32-7.51 (m, 5H, Harom.); 3.69 (q, J=6.6 Hz, 1H, H-1´);
3.54-3.65 (m, J=5.1 Hz, 1H, H-4); 2.90 (q, J=4.2 Hz,
J=9.3 Hz, 1H, H-2); 1.63-1.74 (m, J=4.2 Hz, 1H,
H-3eqv.); 1.42-1.56 (m, J=9.0 Hz, J=4.8 Hz, 1H,
H-3ax.); 1.37 (d, J=6.6 Hz, 3H, H-2´); 1.17-1.35 (m, 6H,
H-5,6,7); 0.84 (t, J=6.3 Hz, 3H, H-8)
(2R,4R,1´R)-4-hydroxy-2-(1´-phenylethylamino)nonanoic
acid (6c)
Yield
0.61g (66%), m.p. 178-180°C
[a ]D26 = 38.0 (c=1,
MeOH)
13C
NMR (NaOD/D2O): 184.8 (C-1); 146.8 (C-1-Ph); 131.6 (C-3,5-Ph); 130.4
(C-4-Ph); 130.2 (C-2,6-Ph); 74.1 (C-4); 63.4 (C-1´); 59.5 (C-2); 42.4 (C-3);
38.9 (C-5); 33.8 (C-6); 27.1 (C-7); 26.3 (C-2´); 24.7 (C-8); 16.2
(C-9)
1H
NMR (NaOD/D2O):
7.30-7.49 (m, 5H, Harom.); 3.69 (q, J=6.9 Hz, 1H, H-1´);
3.54-3.64 (m, J=3.3 Hz, J=13.5 Hz, 1H, H-4); 2.89 (q,
J=5.1 Hz, J=9.0 Hz, 1H, H-2); 1.62-1.73 (m, J=4.5 Hz,
J=3.3 Hz, 1H, H-3eqv.); 1.40-1.55 (m, J=13.8 Hz, 1H,
J=8.7 Hz, H-3ax.); 1.37 (d, J=6.3 Hz, 3H, H-2´);
1.17-1.34 (m, 8H, H-5,6,7,8); 0.85 (t, J=6.3 Hz, 3H,
H-9)
[a ]D26 = 33.7 (c=0.5,
MeOH)
13C
NMR (NaOD/D2O): 185.1 (C-1); 146.8 (C-1-Ph); 131.5 (C-3,5-Ph); 130.3
(C-4-Ph); 130.2 (C-2,6-Ph); 72.4 (C-4); 61.5 (C-1´); 59.3 (C-2); 42.6 (C-3);
38.2 (C-5);29.7 (C-6); 26.3 (C-2´); 24.8 (C-7); 16.2 (C-8)
1H
NMR (NaOD/D2O):
7.30-7.50 (m, 5H, Harom.); 3.71 (q, J=6.6 Hz, 1H, H-1´);
3.60-3.66 (m, J=6.3 Hz, J=11.1 Hz, 1H, H-4); 2.91 (t, J=6.9
Hz, 1H, H-2); 1.63 (t, J=6.9 Hz, J= 12.0 Hz, 2H, H-3); 1.36 (d,
J=6.6 Hz, 3H, H-2´); 1.17-1.35 (m, 6H, H-5,6,7); 0.83 (t, J=6.3
Hz, 3H, H-8)
13C
NMR (NaOD/D2O): 185.1 (C-1); 146.9 (C-1-Ph); 131.5 (C-3,5-Ph); 130.3
(C-4-Ph); 130.2 (C-2,6-Ph); 72.6 (C-4); 61.8 (C-1´); 59.4 (C-2); 42.5 (C-3);
38.4 (C-5); 33.8 (C-6); 27.0 (C-7); 26.2 (C-2´); 24.7 (C-8); 16.2
(C-9)
1H
NMR (NaOD/D2O):
7.25-7.43 (m, 5H, Harom.); 3.71 (q, J=6.3 Hz, 1H, H-1´);
3.59-3.63 (m, J=5.1 Hz, 1H, H-4); 2.92 (t, J=6.9 Hz, 1H, H-2);
1.62 (t, J=6.9 Hz, J=6.0 Hz, 2H, H-3); 1.36 (d, J=6.6 Hz,
3H, H-2´); 1.09-1.28 (m, 8H, H-5,6,7,8); 0.83 (t, J=6.3 Hz, 3H,
H-9)
Acknowledgements.
The authors are grateful to the Slovak Grant Agency for financial support No.
1/6269/99.
References
1. König, W. A.; Haas, W.; Dehler, W.; Fiedler,
H.-P.; Zähner, H.; Liebigs. Ann. Chem. 1980,
622-628.
2. Fehr, T.; Quesniaux, V. F. J.; Sanglier, J. J.;
Oberer, L.; Gschwind, L.; Ponelle, M.; Schilling, W.; Wehrli, S.; Enz, A.;
Zenke, G.; Schuler, J. Antibiot.; 1997; 50,
893-899.
3.Dong, Y.; Pai, N. N.; Ablaza, S. L.; Yu, S.;
Bolvig, S.; Forsyth, D. A.; Le Quesne, P. W. J. Org. Chem. 1999,
64, 2657-2666.
4. Fischer, M. H.; Mrozik, H.; Schoen, W. R.; Shih,
T. L.; Wyvratt, M.; WO 9516692 (1995)
5.Caddick, S.; Jenkins, K.; Chem. Soc. Rev.
1996, 447-456.
6.a) Yamada, M., Nagashima, N., Hasegawa, J., and
Takahashi, S. Tetrahedron Lett. 1998, 39, 9019-9022;b)
Kolarovi?, A.; Berke, D.; Baran, P.; Povaanec, F.; Tetrahedron Letters2001,42, 2579-2582.
7. Miura, T.; Masuo, N.; Fusamae, Y.; Kajimoto, T.;
Ida, Y.; Synlett1998; 631-633.
8. Berke, D.; Kolarovic, A.; Povaanec, F.
Tetrahedron Lett. 2000; 41; 5257-5260.
9. Fischer, J.; Fodor, T.; Dobay, L.; Kiss, B.;
Tóth, G.; Schnatzke, G.; Oetvoes, L. Monatsh.Chem.; 1989;
119; 645-647
10. Jackson, R. F. W.; Wood, A.; Withes, M. J.
Synlett. 1990, 735-736.
11. Steiner, A.; Wessig, P. Helv. Chim. Acta
1996, 79, 1843.
12. Jackson, R. F. W.; Rettie, A. B.; Wood, A.;
Withes, M. J. J. Chem. Soc., Perkin Trans.1 1994,
1719-1726.
13. Jäger, V.; Grund, H.; Buss,,
V.; Schwab, W.; Müller, I.; Schone, R.; Franz, R.; Ehler, R. Bull. Soc. Chim.
Belg. 1983,
9, 1039.
14. Berke, D.; Kolarovi?, A.; Povaanec, F.;
Proceedings of XXVth Conference of Organic Chemists,
Kutná Hora 2001, Czech
Republic.
15. Kametani, T.; Kigasawa, K.; Hiiragi, M.;
Wakisaka, K.; Haga, S. Heterocycles1981, 16,
1205-1242.
16. Yato, M.; Homma, K.; Ishida, A.
Heterocycles 1998, 49, 233-254.
17. Burger, K.; Rudolph, M.; Neuhauser, H.
Liebigs. Ann. Chem. 1991, 1365-1368.
18. Golubev, A. S.; Sewald, N.; Burger, K.
Tetrahedron 1996, 52, 14757-14776.
19. Goldfarb, J.; Fabricnyj, B. P.; Rogovik, V. I.
Izv. Akad. Nauk. SSSR, Ser. Khim. 1963,
2172-2177.
20. Stammers, A.; Burk, M. J. Tetrahedron
Letters 1999, 40, 3325-3328
21. Chapman, K.; Wales, J. S.; Niedzwiecki, L. M.;
Izquiredo-Martin, M. et. el. Bioorg. Med. Chem. L. 1996, 6,
329-332
22. Traber, R.; Hofmann, H.; Loosli, H. R.; Ponelle,
M.; Wartburg, A. von Helv. Chim. Acta 1987, 70, 13-36
23. Hasegawa, H.; Shioiri, N.; Narita, T.; Katori,
T. EP 266 568 (1988)
24. Cicchi, S.; Goti, A.; Brandi, A.; Guarna, A.; De
Sarlo, F. Tetrahedron Lett. 1990, 31,
3351-3354.