Studies towards Dibenzothiophene-S-oxide Arrays and their photochemical Reactivity
Thies Thiemann,a* Kazuya Kumazoe,b Kazuya Arima,b Shuntaro Matakaa
aInstitute of Advanced
Material Study and bGraduate School of Engineering Sciences, Kyushu
University, 6-1, Kasuga-koh-en, Kasuga-shi, Fukuoka 816-8580, Japan
e-mail: [email protected] (for
TT)
Received: 20 August 2001 / Uploaded 21 August 2001
Keywords: Dibenzothiophene-S-oxides, Photoextrusion, Deoxygenation
INTRODUCTION
It is known that
dibenzothiophene-S-oxide can lose oxygen when
photoirradiated.[1-3] The products are the corresponding
dibenzothiophenes. The mechanism of this deoxygenation and the nature of the
released oxygen is still under discussion.[4,5] The authors have
become interested in this field as their original study of the photochemical
behaviour of thiophene-S-oxide showed it to be very dependent on the
substituent pattern of the thiophene-S-oxides[6,7] and that
more than one mechanism (pathway) can operate for one compound. Thus,
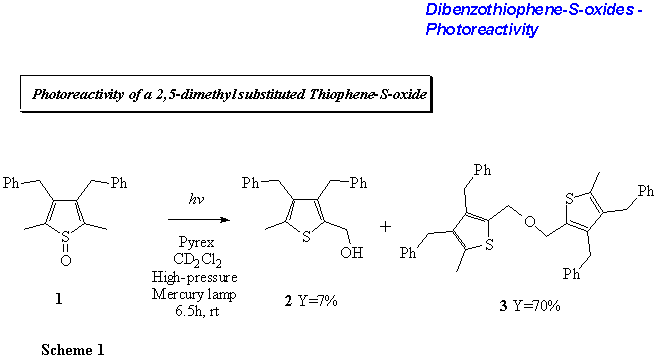
2,5-dimethylthiophene-S-oxide?1 gives hydroxymethylthiophene
2 and bisthienylether 3 upon photoirradiation (Scheme
1).[6] At that time, this seemed to be an indication of a release of
atomic oxygen from the molecule. When substituting the methyl groups for more
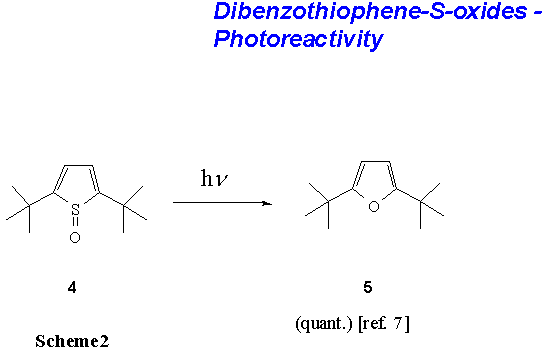
sterically demanding substituents such as tert-butyl as in 4, the
reaction followed a different pattern and the oxygen was introduced into the
heterocyclic ring system ultimately giving the furan 5 as a product
(Scheme 2).[7]
Other compounds, such as
2,3,4,5-tetraphenylthiophene-S-oxide deoxygenate to give the
corresponding thiophenes. This seemed to point towards the possibility of a
bimolecular process in the photo-deoxygenation process, where large substituents
at C-2 and/or C-5 would hinder the formation of an excited
thiophene-S-oxide dimer and would induce the molecule to follow a
different pathway.
As a similar discussion was ongoing in the case of dibenzothiophene-S-oxides (is it a monomolecular or a bimolecular process?), the authors decided to investigate whether a steric hinderance in the neighborhood of the sulfoxy-unit in the dibenzothiophene-S-oxides (i.e., at C-4 and/or C-6) or other changes within the molecule that would be detrimental to a dimer formation would cause a change in the photodeoxygenation behaviour of these compounds. Preliminary results are reported below.
RESULTS AND DISCUSSION

In order to differentiate the C-4 and C-6 positions for multiple coupling reactions it was necessary to functionalize these positions with halo-groups of different reactivity in the Suzuki coupling reaction. For this purpose 4-TMS-dibenzothiophene was lithiated (n-BuLi/THF) and reacted with 1,2-diiodoethane to furnish 4-iodo-6-trimethylsilyldibenzothiophene 12 (yield 67%). Subsequently, 12 was subjected to BTMABr3 (ZnCl2/AcOH) to give 6-bromo-4-iododibenzothiophene 13 (yield 30%, Scheme 3).
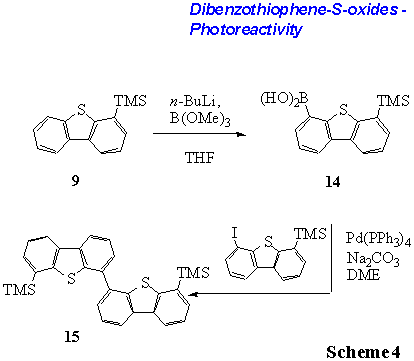
When 4-TMS-dibenzothiophene 9 is
lithiated (n-BuLi/THF) and reacted with trimethylborate
4-TMS-dibenzothiophene-6-boronic acid 14 is obtained (yield 67%, Scheme
4). Here, the trimethylsilyl group at C-4 is used as positional protective
group. As the trimethylsilyl group can exchanged to a halo-substituent (see
above) it can also function as a directing group for a second coupling reaction.
When boronic acid 14 is subjected to Suzuki-Kumada coupling
conditions[11,12] (Pd[PPh3]4,
Na2CO3, DME) the bis-trimethylsilyl-substituted dimer
15 is isolated in 58% yield (Scheme 4).
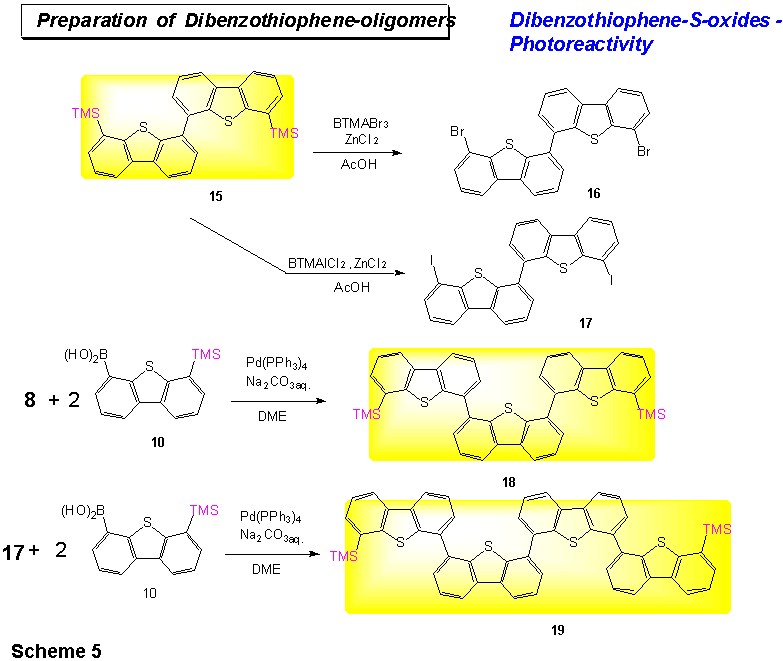
The trimethylsilyl groups in dimer 15 can be exchanged either to bromo-substituents (BTMABr3, ZnCl2, AcOH) to give dimer 16 (yield 97%) or to iodo-substituents (BTMAICl2, ZnCl2, AcOH) to give dimer 17 (yield 97%). When dimer 17 is reacted with 4-TMS-dibenzothiophene-6-boronic acid 14 under Suzuki conditions, the tetramer 6,6-bis-(trimethylsilyl)-4,4-bis(dibenzothiophene) 18 is formed, albeit in low yield (8%, Scheme 5). The low solubility of the symmetric dibenzothiophene-dimer 17 may be the reason (see below).
Trimer 18 can be obtained by
Suzuki-Kumada coupling of 8 and 10 in 13% yield.
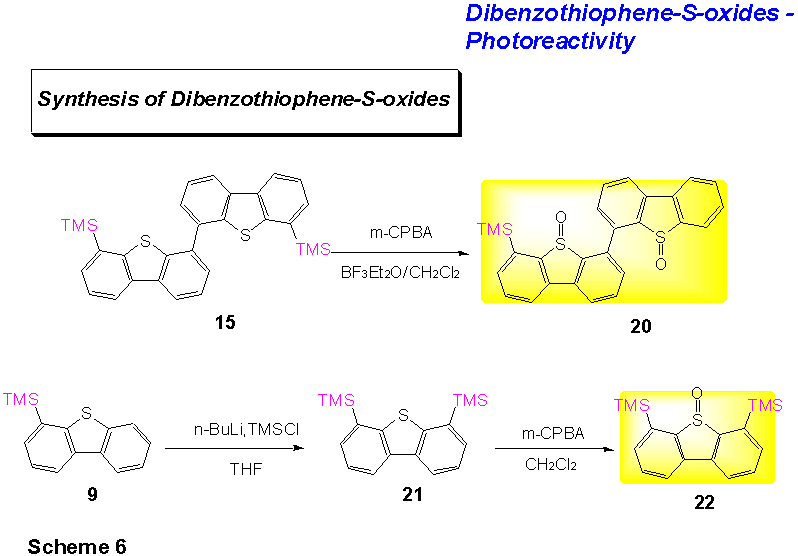
B. Preparation of the Dibenzothiophene-S-oxides. In general, there are various methods for the preparation of dibenzothiophene-S-oxides to dibenzothiophenes.[4] In the following, all dibenzothiophene-S-oxides have been prepared by oxidation with peracids. 4,6-Dimethyldibenzothiophene, which has been prepared by a known method from dibenzothiophene, readily gave 4,6-dimethyldibenzothiophene-S-oxide upon reaction with meta-chloroperbenzoic acid (m-CPBA) in the presence of boron trifluoride etherate (BF3.Et2O) (yield 92%). 4,6-Bistrimethylsilyldibenzothiophene, prepared from 4-trimethylsilyldibenzothiophene 9 [i.) n-BuLi, THF; ii.) TMSCl], was treated with m-CPBA to give the corresponding dibenzothiophene-S-oxide 22 (yield 82%, Scheme 6). When there is a trimethylsilyl group present in the substrate, often it is not advisable to use BF3.Et2O in the reaction. For the most part BF3.Et2O is used in these reactions to activate the peracid and, more importantly, to avoid further oxidation of the S-oxide to the S,S-dioxide by complexation of the Lewis acid to the sulfoxy moiety. The use of BF3.Et2O in trimethylsilyl containing substrates, however, often leads to a cleavage of the trimethylsilyl group (see below).
When bis(6-trimethylsilyldibenzothiophene) 16 is reacted with
m-CPBA in the presence of BF3.Et2O the
mono-TMS-bis(dibenzothiophene-S-oxide) 20 can be isolated (20% yield).
Oxidation of 16 with m-CPBA in the absence of
BF3.Et2O leads to a mixture of products in the
course of the reaction which terminates upon addition of further m-CPBA
with the bis(6-TMS-dibenzothiophene-S,S-dioxide) (yield 46%). A
better approach to bis(6-TMS-dibenzothiophene-S-oxide) may be the
oxidation of the non-symmetric dimeric product 26 or a direct coupling of a
TMS-dibenzothiophene-S-oxideboronic acid such as 25 with
4-iododibenzothiophene-S-oxide 23 (for the preparation, see below).
While dibenzothiophene-S-oxides are
known to be reducible with a number of metals, it nevertheless seemed expedient
to find an access to x-mers of dibenzothiophene-S-oxides by Pd(0)
mediated coupling of halo-substituted dibenzothiophene-S-oxides and of
dibenzothiophene-S-oxide boronic acids. 4-Iododibenzothiophene 7
could be oxidized readily with m-CPBA to
4-iododibenzothiophene-S-oxide 23. Oxidation of
TMS-dibenzothiopheneboronic acid 10 with m-CPBA proved to be more
difficult; also, the corresponding dibenzothiophene-S-oxide 25
could not be obtained in very high purity.
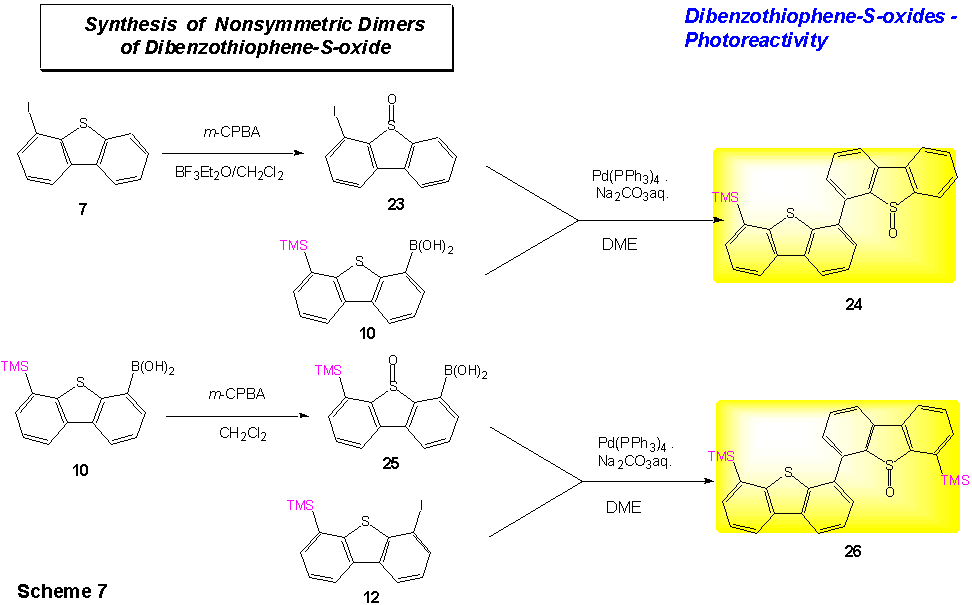
Coupling of 10 and 23 and of 12 and 25 produced the non-symmetric bis(dibenzothiophene)-S-monoxides 24 and 26 (Scheme 7), respectively, in sufficient quantities for initial photodeoxygenation experiments.
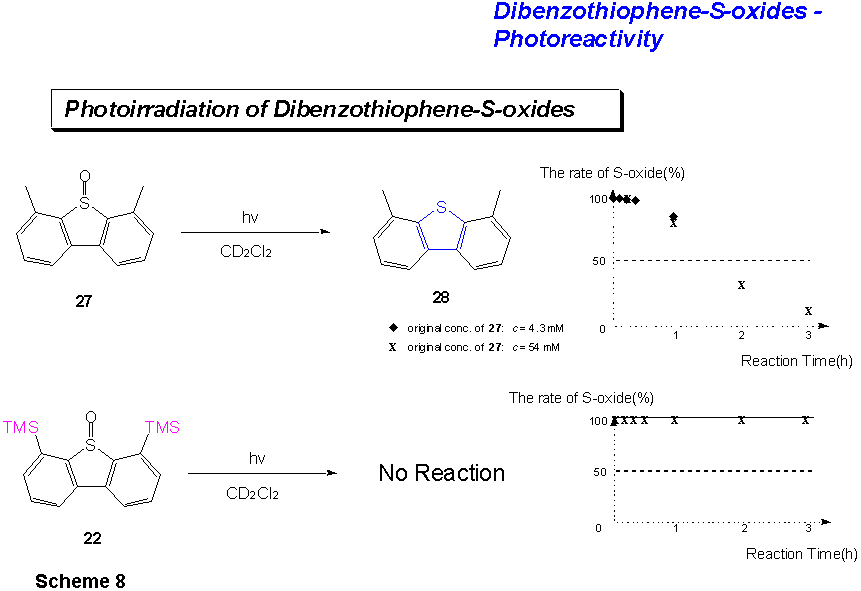
C. Photochemical Experiments with Dibenzothiophene-S-oxides.
4,6-Dimethyldibenzothiophene-S-oxide 27 (54mM) in CD2Cl2 was photoirradiated at 20°C for 12 h. The NMR spectral analysis of the reaction mixture showed 4,6-dimethyldibenzothiophene as the exclusive product. No hydroxylated compound could be observed. Upon purification by column chromatography on silica gel 4,6-dimethyldibenzothiophene 28 could be isolated in 99% yield. The reaction was monitored at 20 min., 1h, 2h and 3h. The experiment was repeated with 4,6-dimethyldibenzothiophene-S-oxide 27 at another concentration (4.3 mM in CD2Cl2). Also in this case, no hydroxylated products could be observed.
4,6-Bis(trimethylsilyl)dibenzothiophene-S-oxide 22 was photoirradiated in CD2Cl2 (c = 4.3 mM) at 20°C for 20h. Here, no photo-product could be obtained. The reaction was monitored at 5 min., 10 min., 20 min., 30 min., 1h, 2h, 3h and 6h. The NMR spectral analysis of the reaction mixture showed exclusively 4,6-bis(trimethylsilyl)dibenzo-thiophene-S-oxide 22.
Also compounds 20 and 26 were photoirradiated in CD2Cl2 at 20°C. The authors supposed that in the case of a possible bimolecular deoxygenation process any structural change that would be detrimental to the formation of a reactive dimer should retard the deoxygenation process.
Similarly substituted monomeric
dibenzothiophene-S-oxides should form dimeric intermediates with greater
ease than the more complex benzothiophene-S-oxide dimers such as
20 or 26.
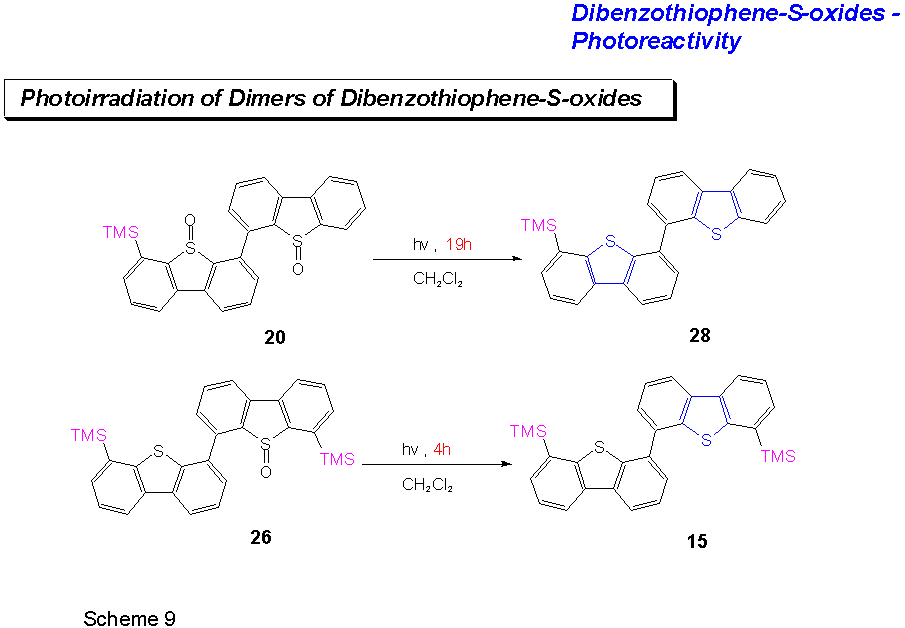
While the photodeoxygenation of 20 was only complete after 14 h (c = 2.1 mM in CD2Cl2), the complete photodeoxygenation of 26 (c = 2.1 mM in CD2Cl2) only took 4 h. 24 has not yet been photoirradiated.
DISCUSSION
In recent years there has been a discussion of whether in the deoxygenation of dibenzothiophene-S-oxides atomic oxygen O(3P) is produced. While the behaviour of O(3P) is well-known in the gas phase,[13,14] irrevocable evidence of O(3P) in solution chemistry has been scarce. It has been suggested, however, that O(3P) is formed when pyridine-N-oxide is irradiated at 308 nm in acetonitrile.[15] The photodeoxygenation of dibenzothiophene-S-oxide has been used in the hydroxylation of interior sites within polyolefinic films.[16] Nevertheless, all authors stress that it is not yet possible to absolutely prove the existance of O(3P) in solution.
In the above experiments the dibenzothiophene-S-oxides were chosen in such a way that their substituents would affect the formation of dimer intermediates due to their steric demand or that the formation of dimer intermediates would be affected by the shapes of the molecules. No optical filter was used in the photoirradiation Thus, the compounds were photoirradiated with light of a broad spectral range (l > 315 nm). All experiments were carried out in CD2Cl2. CD2Cl2 itself can also react with O(3P).
Nevertheless, the following observations were made from the above experiments: A.) 4,6-Dimethyldibenzothiophene-S-oxide 27 cleanly deoxygenates to 4,6-dimethyldibenzothiophene 28. No hydroxylated product can be found. If O(3P) is present in the reaction, then CD2Cl2 competes effectively for it with two well-placed methyl groups. B.) 4,6-Trimethylsilyldibenzothiophene-S-oxide 22 does not deoxygenate at all under the conditions. This can be an indication that in this case a reaction from an excited dimer is necessary for the deoxygenation to take place. C.) The dimeric dibenzothiophene-S-oxides on the whole react more slowly than either dibenzothiophene-S-oxide itself or 4,6-dimethyldibenzothiophene-S-oxide 27. The presence of a dibenzothiophene-unit in the dimeric S-oxide as in 26 enhances the deoxygenation of the compound.
At the moment the photoirradiation experiments are repeated in deuterated benzene and careful kinetic measurements of the deoxygenation process are taken. Furthermore kinetic experiments on the co-photoirradiation of dibenzothiophenes and dibenzothiophene-S-oxides are carried out.
EXPERIMENTAL SETUP
General. For the irradiation a Rikoh-Kagaku-Sangyo RIKO 1KW high-pressure mercury lamp was used. The lamp was in a glass-jacket cooled by water. The sample tubes (pyrex, glass thickness 0.37 mm) were immersed in methanol 7 cm from the lamp (non-rotating). During the photoirradiation the samples were measured by 1H NMR (JEOL EX-270, JEOL JNM-LA 395, or JEOL JNM-LA 600) at the times given in the text. After the photoirradiation, the reaction mixtures were concentrated in vacuo and the residue was subjected to column chromatography on silica gel.
Typical Preparative Example. 20 (5 mg) in deaerated CD2Cl2 (1 mL) was photoirradiated for 19h at rt. A pyrex tube was used. Thereafter, the solvent was evaporated and the residue was subjected to column chromatography on silica gel (ethyl acetate/hexane 1:3) to give 28 (3 mg, 64%); 1H NMR (600 MHz, CDCl3) d 0.62 (s, 9H, SiMe3), 7.36 7.81 (m, 10H), 8.07 8.21 (m, 3H); MS (70 eV) m/z (%) 438 (100). HRMS Found: 438.0933. Calcd. for C27H22SiS2: 438.0932.
REFERENCES