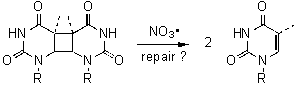
[C0005]
Oxidative Cleavage of Cyclobutane Pyrimidine Dimers by Photochemically Generated Nitrate Radicals (NO3l)
Oliver Krüger and Uta Wille*
Institut für Organische Chemie der Christian-Albrechts-Universität Kiel, Otto-Hahn-Platz 4, 24098 Kiel, Germany
Tel.: +49 +431 880-1179, Fax: +49 +431 880-1558, E-Mail: [email protected]
http://scholle.oc.uni-kiel.de/wi/ewihome.html
Received: 15 August 2001 / Uploaded 22 August 2001
Introduction
The thymine cyclobutane dimer (T<>T) is the most important product formed between two adjacent thymine bases through a [2+2] photo-cycloaddition upon exposure of the DNA to UV radiation. This damage can lead, if not repaired, to mutations or cancerogenous cell growth. The repair of T<>T in bacterials is achieved by the dimer specific repair enzyme photolyase through a direct one-electron reduction followed by cycloreversion of the resulting unstable cyclobutane radical anion and subsequent oxidation to the thymine monomers. It was shown that the cleavage of the cyclobutane pyrimidine dimer could also proceed through an oxidative pathway, since the cyclobutane radical cation is similarly unstable. This oxidative repair was recently used by Barton´s group as a mechanistic probe to examine the long-range charge transfer through the DNA base stack.[1]
During the course of our ongoing study on
the oxidative damage of nucleosides caused by nitrate radicals (NO3l),
we wondered, whether the oxidation power of the electrophilic NO3l
could also have a positive impact on DNA by repairing already existent damages
through an oxidative pathway, e.g. pyrimidine cyclobutane dimers:
Results
As a starting point in our study on the NO3l reaction with pyrimidine cyclobutane dimers, we decided to use simple model compounds, as the stereoisomeric C5-C5´ and C6-C6´-linked N-methylated dihydrouracil and dihydrothymine dimers 2a-d and 4a-b, respectively (Scheme 1).[2]

Scheme 1: Synthesis and stereochemistry of the isomeric pyrimidine cyclobutane dimers 2a-d and 4a-b. The data in parenthesis represent the irreversible half peak anodic potentials Ep/2 in MeCN.[3]
NO3l was in situ generated in an acetonitrile solution of the dimers through photolysis of cerium(IV) ammonium nitrate (CAN) according to (eq 1). These conditions could be considered to simulate the hydrophobic environment in the DNA base stack.
(NH4)2Ce(NO3)6 + hn® NO3l + (NH4)2Ce(NO3)5 (1)
1. NO3l and Uracil Pyrimidine Dimers
The experimental results for the reaction of NO3l with the uracil cyclobutane dimers 2a-d are depicted in Scheme 2. In the first row are shown the products of the NO3l reaction with a mixture of 2a-d containing each isomer in nearly equal amounts. The four rows behind represent the data for the NO3l reaction which the individual uracil dimers.
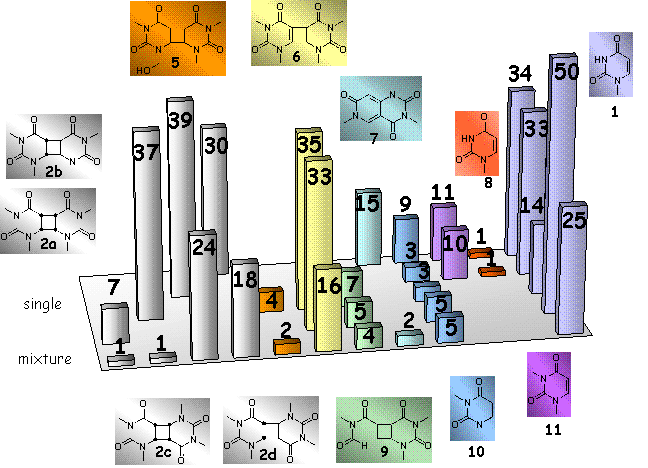
Scheme 2:
GC analysis of the reaction of the reaction of the uracil cyclobutane dimers
2a-d with NO3l.
The data are relative peak areas without an internal standard. Conditions: [2]:[CAN]
= 5, irradiation time = 2 h. Product assignment was performed by GC-MS.
Interestingly, the rate of the NO3l-induced splitting showed a significant dependence on the stereochemistry and the substitution pattern at the cyclobutane ring in 2a-d. Examination of the the data in the first row show that in the mixture of all four isomers the syn isomers 2a and 2b are cleaved faster by NO3l than the anti isomers 2c and 2d.
The data in Scheme 2 show also that cleavage to the monomer 1 did not occur exclusively in the reaction of NO3l with 2a-d, but formation of varying amounts of byproducts 5-11 was also observed. The origin of these compounds was determined by reacting 2a-d separately with NO3l, and their structures were tentatively assigned by GC-MS.
With the exception of the dihydrouracil 10, which is formed in a reaction of the monomer 1 with NO3l and therefore appeared in each reaction under investigation, it is obvious that the product distribution strongly depended on the stereochemistry at the cyclobutane ring in 2a-d:
The C5-C5´linked uracil dimer 6, which is by far the most important by-product, and the formamide 9 were only formed in the reaction of the syn isomers 2a and 2b. But whereas in the case of 2a a preferred splitting to the monomer 1 and minor formation of 6 was observed, the latter is the major product in the reaction of NO3l with 2b, and cleavage into 1 is only a minor reaction pathway.
The NO3l reaction with 2b lead also to formation of the hydroxylated species 5.
The N-dealkylated uracils 8 and 11, as well as the bicyclic compound 7 exclusively appeared, although in only minor amounts, in the reaction of the anti isomers 2c and 2d with NO3l. However, both of these dimers are, like 2a, cleaved to the monomer as the major reaction pathway.
Mechanism
Both electron transfer and hydrogen atom abstraction by NO3l may be considered as the primary step in the reaction with 2a-d. The additional finding of a slow CAN-induced cleavage of 2a suggests that electron transfer should also occur in the reaction with NO3l, since the oxidation potential of the latter is significantly higher than that of CAN. Competion experiments, where equal amounts of N(1),N(3)-dimethylthymine 3 and the dimer 2a and 2c, respectively, were irradiated in the presence of 0.5 equivalents of CAN, showed that 2a,c and 3 react with rates of the same order of magnitude. However, it turned out that 2a is more reactive than 2c and even than 3 Since the reaction of 3 with NO3l is expected to proceed through an initial electron transfer,[4] we therefore believe that this holds true also for the NO3l-induced splitting of the dimers 2a-d.
Based on this, a mechanism could be proposed, which is shown in Scheme 3 for the exemplary reaction of NO3l with the syn isomers 2a,b.
Initial electron transfer at N(1) adjacent to the cyclobutane ring would lead to the radical cation 12. This species could loose an aminomethyl unit to give the the formamide 9. Deprotonation at the N(1)-methyl group in 12 would lead to radical 14, which could be expected to be the precursor for the hydroxylated species 5 through further reactions with NO3l.
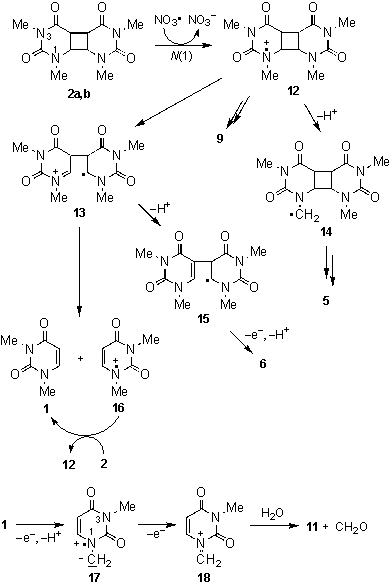
Scheme 3: Proposed mechanism.
Formation of the monomer 1 is believed to proceed stepwise from 12 by first splitting of the C6-C6´ bond to give 13, followed by cleavage into 1 and its radical cation 16. The latter could initiate a radical chain through oxidation of further dimer molecules 2. The suggestion of a radical chain in the present system is based on the finding that consumption of 2a-d was always significantly higher than could be expected from the ratio [2a-d]:[CAN] in the starting reaction mixture (Table 1). The C5-C5´ linked dimer 6 might be formed in competition to the splitting reaction by elimination of a proton in 13, followed by further oxidation and deprotonation of the radical intermediate 15.
The NO3l induced splitting of the anti isomers 2c,d is expected to proceed principally in the same way, although the products and their distribution are different. Their lower reactivity should be due to their higher oxidation potentials (see Scheme1). In an independent experiment it was veryfied that the N-dealkylated uracils 8 and 11 result from a reaction of 1 with NO3l. The dealkylation is expected to proceed through a stepwise electron transfer and deprotonation sequence [shown in Scheme 3 for the dealkylation at N(1)] and finally hydrolysis of the imminium species 18 by trace amounts of water, which are present in the system due to the hygroscopic CAN. However, because of the present uncertainty in the structure assignment for compound 7, we prefer not to speculate about the reaction pathway leading to its formation.
2. NO3l
and Thymine Pyrimidine Dimers
Of the four possible stereoisomers of the thymine cyclobutane dimer only the cis-configurated compounds 4a,b were obtained in satisfactory yield and purity. It turned out that by reaction with NO3l the cis-syn isomer 4a was cleaved to the monomer 3 at a faster rate than the cis-anti isomer 4b (Scheme 4, left side). This finding is in accordance with the results of the NO3l-induced cleavage of the uracil cyclobutane dimers 2a-d (see Scheme 2). The anti isomers possess higher redox potential, which is ca. 0.35- 0.40 V higher than that of syn isomers (Scheme 1).
Competition experiments, in which equal amounts
of the cis-syn isomers 2a and 4a were reacted with
0.5 eq. of NO3l
revealed that the uracil dimer 2a is cleaved significantly faster than
its thymine derivative 4a (Scheme 4, right side).

Scheme 4:
GC analysis of the reaction of the reaction of the uracil cyclobutane dimers
4a,b with NO3l.
This behavior cannot be due to the ease of the initial electron transfer step, because the redox potentials of both species 2a and 4a are practically identical (see Scheme 1). We believe that sterical hindrance may be of significant importance in the electron transfer induced splitting reaction. With respect to the uracil dimer 2a the cyclobutane ring in the thymine dimer 4a is significantly more crowded by the two additional methyl groups.
As in the NO3l-induced
splitting of the uracil dimers 2a-d also in the reaction of
4a,b with NO3l
formation of byproducts, but, however, to a lesser extent, was observed. These
byproducts were not yet identified, but, interestingly, a dependence of their
formation on the stereochemistry at the cyclobutane ring was evident.
Conclusions
To conclude, we have demonstrated that the
strongly oxidizing NO3l
is able to cleave the cyclobutane dimers 2a-d and 4a,b
to the monomers 1 and 3, respectively, through an initial electron
transfer step. The rate of the splitting reaction and the splitting efficiency,
as well as the distribution of the by-products formed besides the monomer 2
and 4 shows a significant dependence on the stereochemistry and the
substitution pattern at the cyclobutane ring. However, we do not know the reason
for this behavior yet. Therefore, extensive studies on the reaction mechanism of
the NO3l
induced splitting of these as well as of further pyrimidine cyclobutane dimers
are in current progress in our laboratory.
Acknowledgment
Financial support by the Deutsche
Forschungsgemeinschaft, Dr.-Otto-Röhm-Gedächtnisstiftung and Fonds der
Chemischen Industrie is gratefully acknowledged.
References
[1] (a) Vicic, D.A.; Odom, D.T.; Núñez, M.E.; Gianolio, D.A.; McLaughlin, L.W.; Barton, J.K. J. Am. Chem. Soc. 1999, 122, 8603. (b) Dandliker, P.J.; Homlin, R.E.; Barton, J.K. Science 1997, 275, 1465. (c) Dandliker, P.J.; Núñez, M.E.; Barton, J.K. Biochemistry1998, 37, 6491.
[2] Krüger, O.; Wille, U. Org. Lett. 2001, 3, 1455.
[3] Pac. C.; Kubo, J.; Majima, T.; Sakurai, H. Photochem. Photobiol. 1982, 36, 273.
[4] Krüger, O.; Wille, U.; Steenken, S. to be published.