Peter Sawatzki, Matthias Skovron, Gerhild van Echten-Deckert, and Thomas Kolter*
Kekulé-Institut für Organische Chemie und Biochemie der Universität, Gerhard-Domagk-str.1, 53121 Bonn, Germany. *Phone: Int / 228 / 73 57 98, Fax: Int / 228 / 73 77 78, E-mail: [email protected]
Received: 15 August 2001 / Uploaded 22 August 2001
Abstract: We report on the synthesis of two ceramide analogs, and their effect on de novo sphingolipid biosynthesis. 1-Deoxy-1-fluoroceramide has been prepared starting from D-galactose in 0.8 % yield. Bioisosteric replacement of the amide-group by a sulfonamide led to an additional target molecule, which has been synthesized also from D-galactose in 5.1% yield. Initial results in cultured murine neurons indicate a competition of the ceramide derivatives with endogenous by synthesized ceramide.
Keywords: Ceramide, Bioisosteric Replacement, Metabolism
1. Introduction
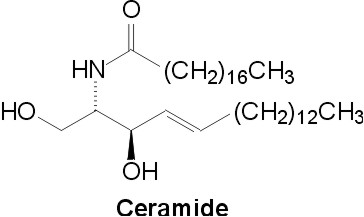
Fig. 1. Ceramide structure
Ceramide (N-Acyl-D-erythro-sphingosine, Fig. 1) is a structural component of mammalian glycolipids and of a phospholipid, sphingomyelin [1]. Ceramide derivatives attracted attention as potential therapeutic agents for the treatment of cancer, allergy, and other diseases originating from cell regulation disorders [2]. Ceramide itself is a signaling substance that can be released from sphingomyelin in response to extracellular and intracellular stimuli. In general, it mediates antimitogenic cellular effects like differentiation, apoptosis, or cellular senescence [3]. Details of this pathway including the identity of the presumed ceramide binding proteins are not entirely clear. One reason for this is the metabolic coupling of ceramide with other bioactive lipids like ceramide-1-phosphate, sphingosine, and sphingosine-1-phosphate. For investigation and triggering ceramide metabolism and signaling, we are interested in ceramide analogs and derivatives of enhanced metabolic stability.
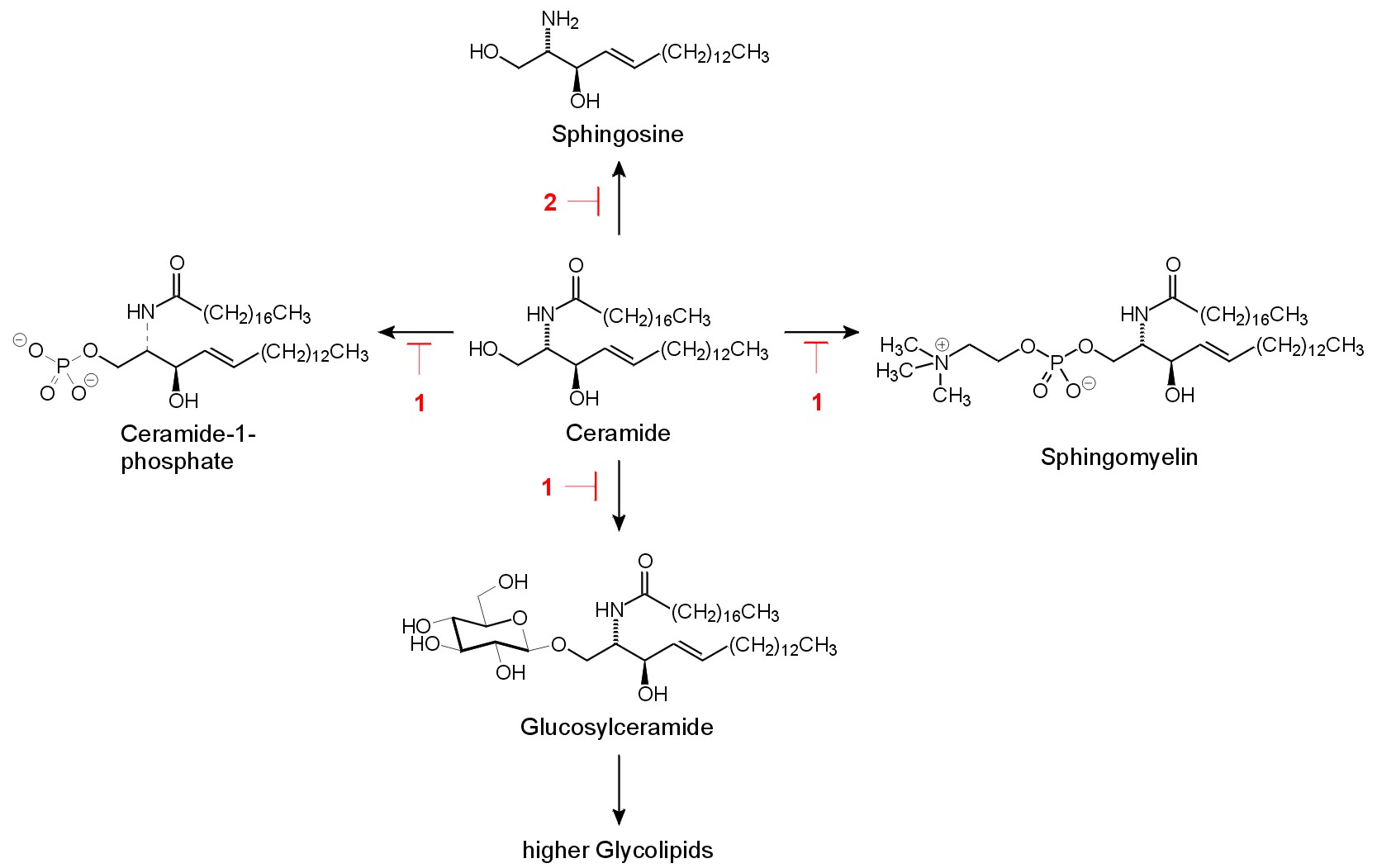
Fig. 2. Ceramide metabolism. Potential inhibition sides by the target compounds 1 and 2 are indicated.The major metabolic reactions of exogenous [4] and endogenous [5] ceramides are the formation of sphingomyelin and glucosylceramide (Fig. 2). Both reactions require the presence of the 1-OH group of ceramide. A modification that prevents formation of sphingomyelin, glucosylceramide, but also of ceramide-1-phosphate and galactosylceramide is the replacement of the 1-hydroxyl group by a suitable substitute. Earlier studies indicated that this is possible without eliminating the signaling properties of the lipid: dihydroceramide analogs bearing an alkyl residue instead of the 1-hydroxyl group show suitable properties in different lines of cultured cells [6],[7].
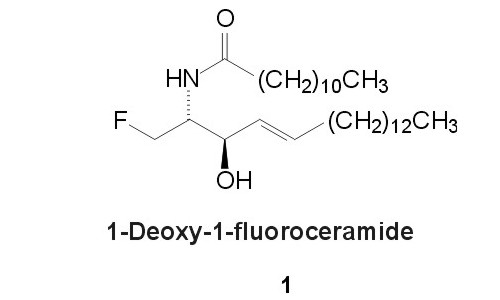
Fig. 3. 1-Deoxy-1-fluoroceramide 1Consideration of the bioisosteric replacement of a hydroxyl group by fluorine (see [8] for review) led us to the design of 1-deoxy-1-fluoroceramide 1 (fig. 3).
Besides metabolic elongation of 1-OH-group, ceramide can also be degraded by several ceramidases to sphingosine. These enzymes of different subcellular localization, cofactor requirement, and pH-optima, cleave the amide linkage within the lipid.
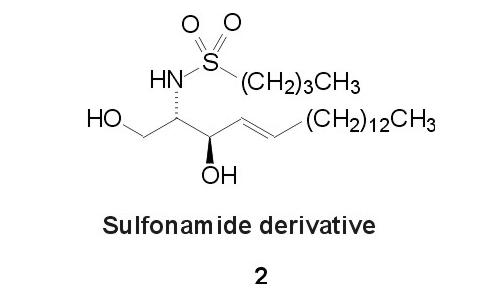
Fig. 4. Sulfonamide analogue 2
Replacement of the amide moiety by a sulfonamide-group is one possibility to enhance the metabolic stability of the parent compound [9]. We attempted to achieve enhanced catabolic stability by incorporation of this modification into the ceramide structure, leading to 2 (Fig. 4). This should also prevent formation of sphingosine-1-phosphate which is an additional signalling substance metabolically derived from ceramide [1].We report the synthesis of 1 and 2 as ceramide analogues of enhanced metabolic stability with high similarity to the parent lipid.
2. Results
Fig. 5. Undesired reaction of DAST with sphingosine derivatives
Various synthetic routes to ceramide, or its preparative precursor, sphingosine, have been reported [10]. Many of them lead to protected long chain 2-tert.-butyloxycarbonylamino-1,3-diols. Reaction of urethane-protected b-aminoalcohols, however, with diethylaminosulfur trifluoride (DAST) leads to heterocyclic products instead of a fluorine for hydroxyl exchange [11] and also in our hands, N-acyl- and N-urethane-protected sphingosine derivatives gave not the desired 1-fluoro-derivatives on reaction with DAST (Fig. 5).

Fig. 6. Azidosphingosine strategy to the fluoro derivative 1
Therefore, we decided to apply the following strategy (Fig. 6). Starting point is the MPM (= p-methoxyphenylmethyl) -protected azidosphingosine 3 which is accessible from Diethyl-D-tartrate [12], but is more conveniently prepared from D-galactose according to a modification [13] of the procedure by Zimmermann and Schmidt [14].

Fig.7. Route to azidosphingosine derivative 7The preparation of this starting material is summarized in Fig. 7. Replacement of the 1-hydroxyl group in 3 by fluorine is achieved with DAST [15] (Fig. 6). The moderate yields in this key step are accompanied by recovery of considerable amounts of starting material 3. The fluorinated azidosphingosine building block 4 is deprotected to 5 using Ceric ammonium nitrate [16] in good yield. Staudinger reaction of 5 with triphenylphosphine in THF / water [17] leads to 1-deoxy-1-fluorosphingosine 6 [18]. 6 can be selectively acylated with lauroyl chloride in methanol in the presence of triethylamine at low temperatures [19] to yield the target compound 1.
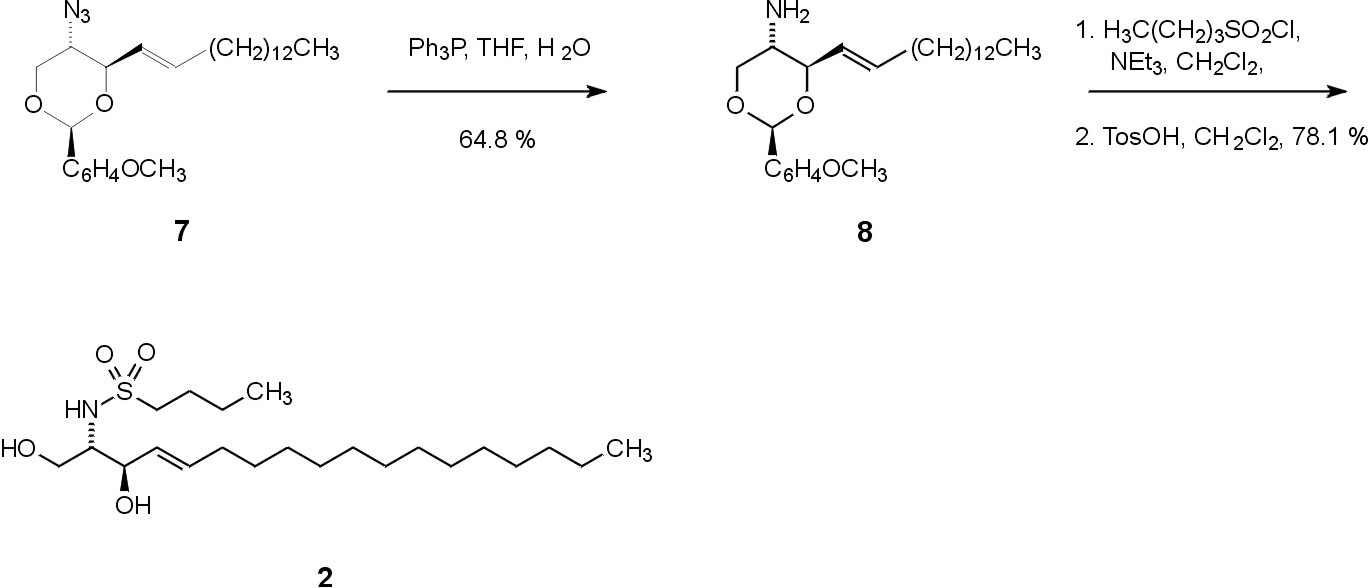
Fig. 8. Synthesis of the sulfonamide derivative 2The synthesis of 2 starts from compound 7, which is also available from D-galactose (Fig. 7). Staudinger reduction of the azide, acylation with an appropriate sulfuryl chloride, and the subsequent deprotection under acidic conditions with toluenesulfonic acid leads to the target compound 2 (Fig. 8).
We investigated the ceramide analogs 1 and 2 on their influence on the incorporation of a biosynthetic precursor, L-serine, into ceramide-containing lipids of primarily cultured cerebellar cells of mice.
In brief [20], the cells were treated with 10, 25, 50, and 100 µM of the target compounds 1 and 2 for 24 h. Radiolabelled 3-[14C]serine was added to the culture medium and incorporation into newly synthesized sphingolipids was analyzed after 24h labelling. Lipids were extracted, separated by thin layer chromatography, and visualized with the aid of a phosphoimager. Radioactivity found in the selected lipids were expressed in relation to untreated cells (control = 100 %). The results of two independent experiments are given in Fig. 9 and 10.
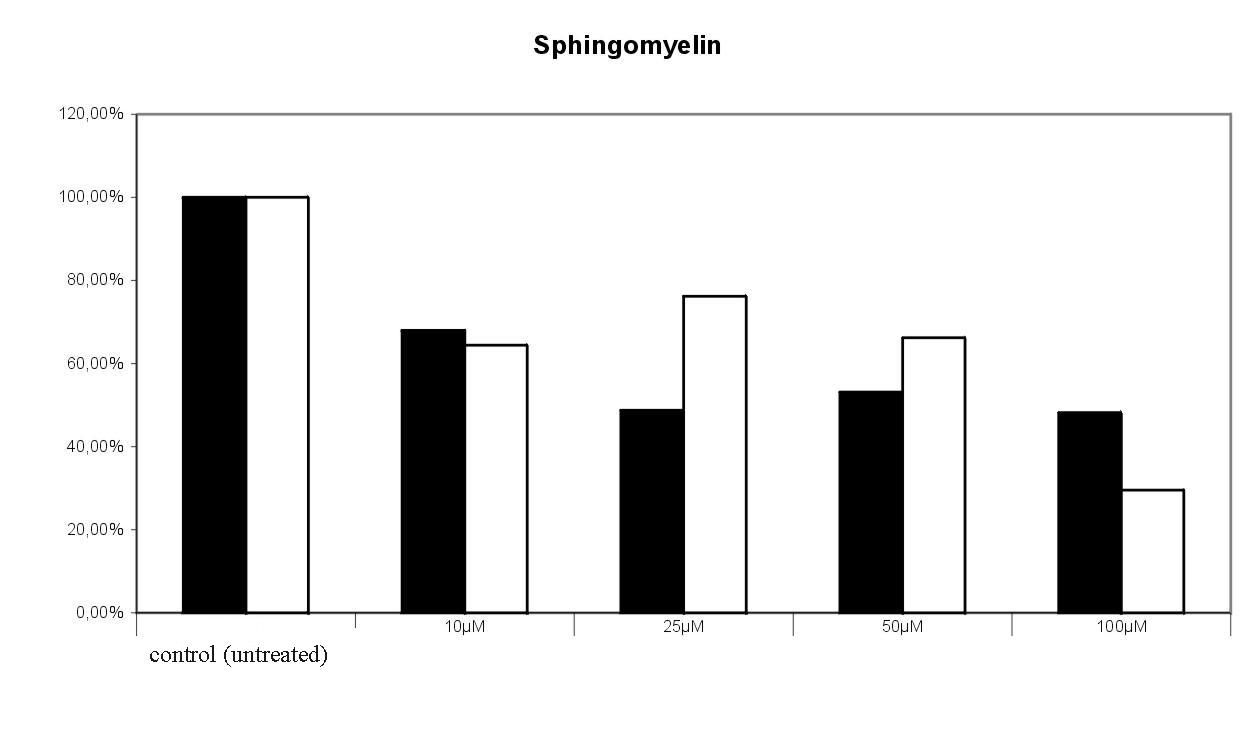
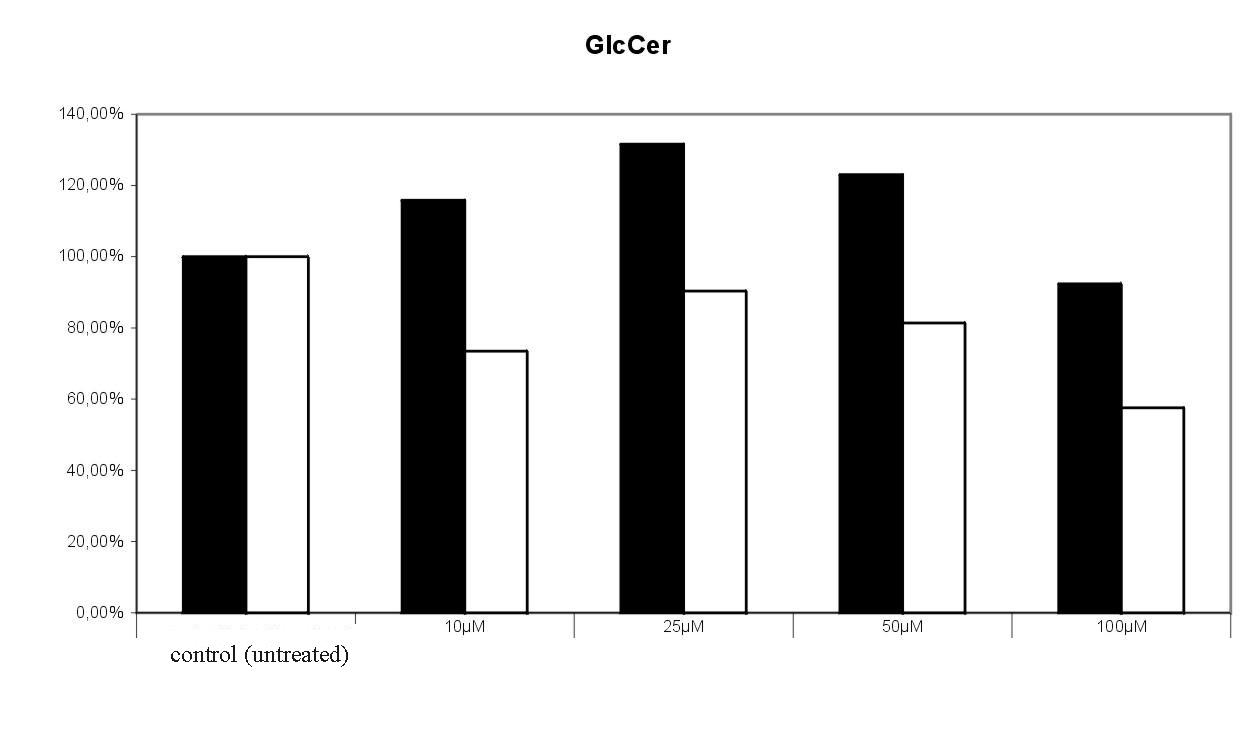
Fig. 9. Effect of 1-deoxy-1-fluoroceramide 1 on the incoroporation of 3-[14C]serine into glucosylceramide and sphingomyelin. Radioactivity found in the tlc spots of untreated cells were set equal 100%. The results of 2 experiments were given , in which the cells were treated with bwith varying concentrations of the target compound. 100 % = 4768 cpm.1-deoxy-1-fluoroceramide 1 led to a concentration dependent decrease of labelled sphingomyelin. Surprisingly, the level of glucosylceramide seems not to be significantly influenced by 1 (Fig. 9).
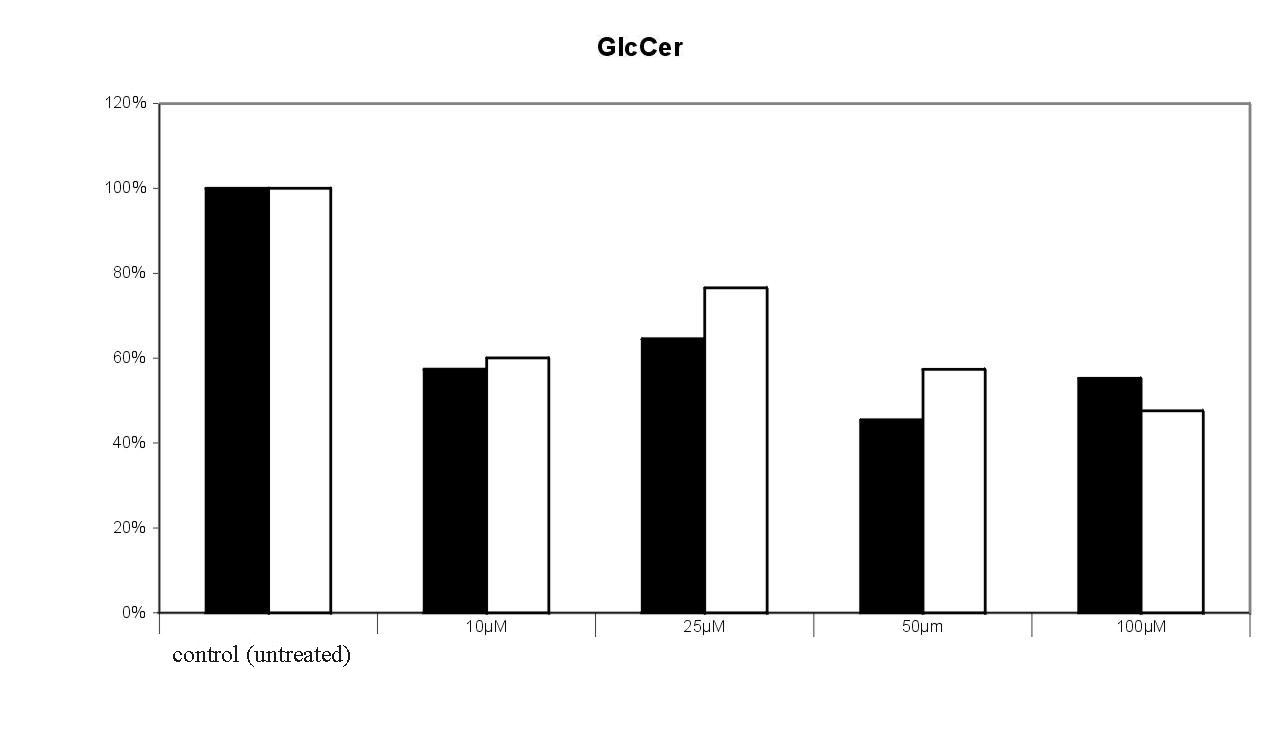
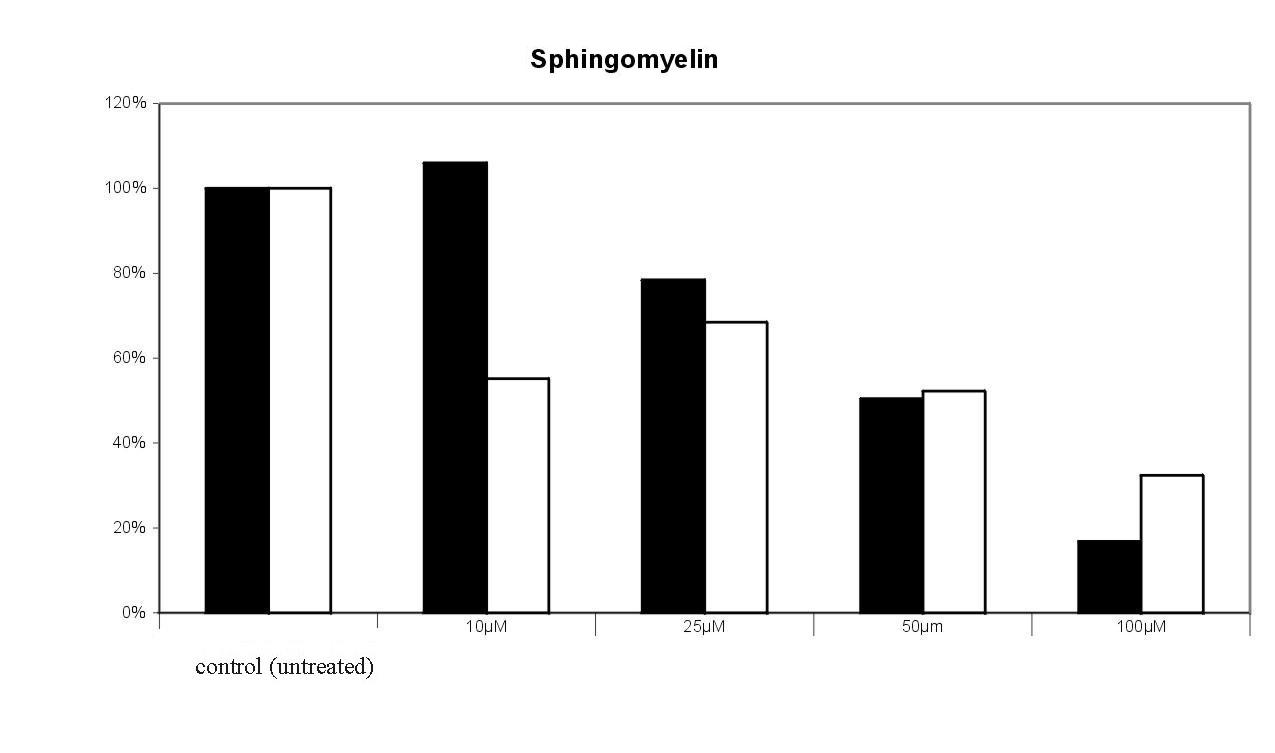
Fig. 10. Effect of the sulfonamide derivative 2 on the incoroporation of 3-[14C]serine into glucosylceramide and sphingomyelin. Radioactivity found in the tlc spots of untreated cells were set equal 100%. The results of 2 experiments were given , in which the cells were treated with bwith varying concentrations of the target compound. 100 % = 7570 cpm.The dose dependent effect of 2 on 3-[14C]serine incorporation into sphingomyelin and glucosylceramide is shown in Fig. 10. Incorporation of radioactivity into downstream metabolites of ceramide is significantly decreased in a dose dependent manner. Sphingomyelin formation was reduced to 25 % by 100 µM of 2. The decrease of glucosylceramide is not as drastic as in the case of sphingomyelin; the de novo synthesis is reduced to 40 50 % by 100 µM of 2. Also the levels of lactosylceramide (to 40 %) and of the higher gangliosides GT1b (up to 40%) and GD1a (up to 20 %) are decreased compared to the control (data not shown).
3. Discussion
The described method provides access to ceramide analogues 1 and 2 and also to the sphingosine derivatives 5 and 6 (fig. 6). The present approach provides access to the 1-fluoro-analog of ceramide in its natural occurring configuration and with the 4,5-trans-double bond which is required for the signaling properties of ceramide [21]. The synthesis presented here makes use of slightly modified intermediates commonly used in the preparation of glycolipids [14] and avoids the unfavorable [11] reaction of urethane- or acyl-protected b-amino alcohols with DAST. An alternative approach leading to 5 has been reported starting from (R)-fluoroalanine [18], which in turn is prepared from (S)-serine [22].
Ceramide analogs with a fluoro-substituent in 3-position have already been prepared. They show apoptogenic properties [23] and have been investigated as inhibitors of protein kinase C [24]. The bioisosteric hydroxyl to fluorine replacement and the substitution of the amide to sulfonamide within the ceramide structure constitutes a valuable addition to the known sphingolipid analogues and derivatives [25].
The initial results of cell culture studies in murine cerebellar neurons indicate a competition of the synthetic derivatives with the endogenously formed parent compound. As can be expected from Fig. 2, the steady state levels of the downstream metabolites were decreased. Due to the low absolute radioactivity found in the ceramide bound, large derivation were observed in the presence of 1 and 2 between the two experiments. It can be concluded that GlcCer synthase shows a higher substrate selectivity than sphingomyelin synthase. Surprisingly, the fluoro substitution is less active towards GlcCer synthase than expected. Albeit further studies are necessary to confirm the enhanced metabolic resistance of 1 and 2, their application to living cells should lead to elevated concentrations of endogenous ceramide. Elevation of this proapoptotic lipid in humans e. g. achieved by intake of nonsteroidal antiinflammatory drugs, seems to lead to protection from colon cancer [26]. In general, several attempts have been made to pharmacologically increase ceramide levels for cancer treatment [27]. Our approach utilizes the bioisosteric replacement for enhancement of metabolic stability.
4. Acknowledgment
We thank Prof. Dr. K. Sandhoff, Bonn, the Deutsche Forschungsgemeinschaft (SFB 284) and the Fonds der Chemischen Industrie for the support of this work, J. Hörnschemeyer for recording the MALDI mass spectra, U. Damen for IR-spectra, U. Weynand and C. Schmidt for the NMR spectra.
5. References
[1] T. Kolter, K. Sandhoff, Angew. Chem. 1999, 111, 1633 - 1670, Angew. Chem. Int. Ed. 1999, 38, 1532 - 1568.
[2] T. Sakai, Y. Koezuka, Exp. Opin. Ther. Patents 1998, 8, 1673 - 1682.
[3] Y. A. Hannun, C. Luberto, K. M. Argraves, Biochemistry 2001, 40, 4893 4903.
[4] N. D. Ridgway, D. L. Merriam, Biochim. Biophys. Acta 1995, 1256, 57 - 70.
[5] G. van Echten-Deckert, A. Klein, T. Linke, T. Heinemann, J. Weisgerber, K. Sandhoff, J. Lipid Res. 1997, 38, 2569 - 2579.
[6] T. Wieder, C. C. Geilen, T. Kolter, F. Sadeghlar, K. Sandhoff, R. Brossmer, P. Ihrig, D. Perry, C. E. Orfanos, Y. A. Hannun, FEBS Lett. 1997, 411, 260 - 264.
[7] M. Bektas, Y. Dullin, T Wieder, T. Kolter, K. Sandhoff, R. Brossmer, P. Ihrig, C. E. Orfanos, C. C. Geilen, Exp. Dermatol. 1998, 7, 342 - 349.
[8] a) G. A. Patani, E. J. LaVoie, Chem. Rev. 1996, 96, 3147 3176; b) D. O'Hagan, H. S. Rzepa, J. Chem. Soc. Chem. Commun.1997, 645 - 652.
[9] D. B. A. de Bont, K. M. Sliedregt-Bol, L. J. F. Hofmeyer, R. M. J. Liskamp, Bioorg. Med. Chem. 1999, 7, 1043 1047.
[10] P. M. Koskinen, A. M. P. Koskinen, Synthesis1998, 1075 - 1091.
[11] H. Zhao, A. Thurkauf, Synlett 1999, 1280 - 1282.
[12] P. Somfai, R. Olsson, Tetrahedron 1993, 49, 6645 - 6650.
[13] K. Clasen, A. Giannis, unpublished.
[14] P. Zimmermann, R. R. Schmidt, Liebigs Ann. Chem.1988, 663 - 667.
[15] J. Middleton, J. Org. Chem. 1975, 40, 574 - 578.
[16] R. Johansson, B. Samuelsson, J. Chem. Perkin Trans. I 1984, 2371 - 2374.
[17] Reaction conditions according to S. Nagarajan, B. Ganem, J. Org. Chem. 1987, 52, 5044 - 5046.
[18] A. Kozikowski, J. Wu, Tetrahedron Lett. 1990, 31, 4309 - 4316.
[19] Acylation conditions: L. Elsen, unpublished.
[20] G. van Echten-Deckert, A. Zschoche, T. Bär, R. R. Schmidt, A. Raths, T. Heinemann and K. Sandhoff, J. Biol.Chem. 1997, 272, 15825 15833.
[21] A. Bielawska, H. M. Crane, D. Liotta, L. M. Obeid, Y. A. Hannun, J. Biol. Chem. 1993, 268, 26226 - 26232.
[22] P. J. Reider, R. S. E. Conn, P. Davis, V. J. Grenda, A. J. Zambito, E. J. Grabowski, J. Org. Chem. 1987, 52, 3326 - 3334.
[23] S. De Jonghe, I. van Overmeire, J Gunst, A. De Bruyn, C. Hendrix, S. van Calenbergh, R. Busson, D De Keukeleire, J. Philippé, P. Herdewijn, Bioorg. Med. Chem. Lett. 1999, 9, 3159 - 3164.
[24] S. De Jonghe, I. van Overmeire, S. Poulton, C. Hendrix, R. Busson, S. van Calenbergh, D. De Keukeleire, S. Spiegel, P. Herdewijn, Bioorg.Med. Chem. Lett. 1999, 9, 1375 - 3180.
[25] R. Ghidoni, G. Sala, A. Giuliani, Biochim. Biophys. Acta 1999, 1439, 17 - 39.
[26] T. A. Chan, P. J. Morin, B. Vogelstein, K. W. Kinzler, Proc. Natl. Acad. Sci. USA 1998, 95, 681 686.
[27] N. S. Radin, Eur. J. Biochem. 2001, 268, 193 204.