Fifth International Electronic Conference
on Synthetic Organic Chemistry (ECSOC-5), http://www.mdpi.org/ecsoc-5.htm, 1-30
September 2001
[E0007]
Solid Phase Carbonylation in Fast Microwave-Mediated Combinatorial Drug
Discovery

Nils-Fredrik K.
KAISER,

Anders
HALLBERG
and

Mats LARHED
[*]
Uppsala University,
BMC, Department of Organic Pharmaceutical Chemistry,
P.O. Box 574, SE-751 23 UPPSALA,
SWEDEN.
Fax: +46 (0)18 471 4474, e-mail: mailto:[email protected]
Received: 15 August 2001 / Uploaded 22 August 2001
Drug Development
Today
With the recent
development of molecular biology and genetics, the bottleneck in drug discovery
has shifted from target identification to the development of candidate
drugs[1]. The techniques of combinatorial chemistry have evolved to
meet this demand of rapid and diverse substance production, by focusing on
automated handling of a large number of samples rather than on experimental
methodologies.[2] For those combinatorial synthesizers available
where gases can be distributed via a common and open link, contamination between
the different reaction wells constitutes an immediate risk. The construction
of fast, reliable and convenient gaseous protocols for chemistry is therefore
important for the chemical development of combinatorial
chemistry.
Mo(CO)6 as Carbonylating Agent -
Development [3]
The finding that
Mo(CO)6 under certain conditions of fast microwave heating
demonstrated a controlled liberation of carbon monoxide encouraged us to
investigate the construction of a new convenient synthetic strategy for
combinatorial carbonylations where carbon monoxide was generated
in-situ.
By heating a mixture of
an aryl bromide or aryl iodide (1 equiv.), an amine (1.2 equiv.),
Mo(CO)6 (0.33 equiv.), a palladium catalyst, K2CO3
(aq) as base and diglyme as solvent in a closed vessel with microwaves,
amides were formed in medium to high yields (65 - 85 % isolated, Table 1, Figure
1).
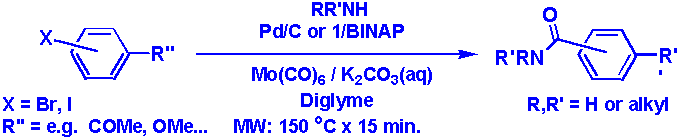
Figure 1:
Conditions for the palladium-catalyzed amidation utilizing in-situ
generated carbon monoxide from Mo(CO)6
Iodides could be
coupled with solid Pd/C as catalyst, while bromides required a homogeneous
catalyst. Thus, for aryl iodides an extremely simple workup by extraction with
2M HCl(aq) and diethyl ether, followed by filtration, afforded the pure amides.
Among the homogeneous catalysts tested for aryl bromides, the most suitable to
use was the deep red 2:1 mixture of BINAP and Herrmanns catalyst (1)
dissolved in toluene.
Table 1: Carbonylations
according to figure 1 employing the in-situ CO generation
protocol.
|
Entry |
R |
Reagent
(RRNH or
other) |
Yield |
|
1
2
3
4
5
6
7
8
9 |
MeO-
Me-
F3C-
Ac-
MeO-
Me-
F3C-
Ac-
Me |
n-BuNH2
n-BuNH2
n-BuNH2
n-BuNH2
Piperidine
Piperidine
Piperidine
Piperidine
Water |
68
[a]
72
[a]
78
[a]
79
[a]
65
[a]
68
[a]
75
[a]
76
[a]
87
[a,b] |
| [a]: Average isolated yields from 2-6 runs.
Aryl iodides and bromides produced essentially the same yields, although
Pd/C was used for the iodides while 1/BINAP was used for the
bromides. [b]: Carboxylic acid. Ethylene glycol was added as co-solvent,
making up ca. 30 % of the
solvent. |
Interestingly, the
carbonylations could also be performed in open vessels without any loss in
yield, suggesting that enough carbon monoxide is dissolved in the reaction
mixture to furnish complete conversion.
The addition of
ethylene glycol as co-solvent to the diglyme/K2CO3(aq)
mixture resulted in competitive formation of the corresponding benzoic acid
derivative instead of the amide (Table 1, entry 9). The omission of amine made
this procedure a powerful tool for making the corresponding benzoic acid from an
aryl halide.[4]
Library Synthesis of HIV-Protease Inhibitors -
Application
An efficient HIV-drug
must ideally have a therapeutic activity not only on the native forms of HIV but
also on its mutants. Inhibition of native HIV-1 protease has been demonstrated
by the C2-symmetric structure 2.[5] A recent
discovery demonstrates that infected patients prescribed with the HIV-drug
Ritonavir™ evolve specific mutations in the protease active site.
These mutations are situated very close to the phenyl rings of 2, within
its enzyme-inhibitor complex. Therefore, the synthesis of aryl analogues to
2 was attractive in our search for new HIV-drugs.
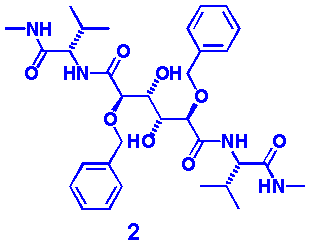

Figure 2:
The application of the carbonylation protocol for rapid production of
HIV-protease inhibitors.
By using halogenated
analogues of 2 (meta, meta dibromo (3) and ortho, ortho diiodo
(4)) in combination with the carbonylation protocol described above, a
focused department stock-room library of amide-derivatives was synthesized
(Figure 2). Generation of the library was undertaken employing the microwave
heating Smith Synthesizer™. Rapid synthesis and repeated
mass-triggered LC-MS purification resulted in a 16 % library yield of 36
isolated compounds. The crude reaction mixtures contained mono- and di-amidated
inhibitors as well as unreacted starting material, where the contribution of the
mono-amidated product varied from 0 30 % of the total yield. Amine 1 10
(Figure 3) are some of the amines utilized.

Figure 3:
Different amines employed in the synthesis of a small sized department
stock-room library of HIV-1 protease inhibitors.
Test results on native
HIV-1 protease indicate that compounds of the ortho- and meta-substituted
structure type might be potent and mutant resistant inhibitors.
& References:
[*] [1] Candidate drugs are new
substances that have a beneficial therapeutic activity towards a disease and a
commercial potential.
[2] a) G. Jung, Combinatorial Chemistry, Whiley-VCH, Weinheim,
Germany, 1999; b) R. E. Dolle, K. H. Jr. Nelson, J. Comb. Chem.
1999, 1, 235.
[3] N-F. K. Kaiser, A. Hallberg,
M. Larhed, In-Situ Generation of CO from M(CO)x, A Convenient
& Fast Route to Pd-catalyzed Amidations. Submitted.
[4] For other similar
approaches to the synthesis of benzoic acids see: a) Y. Masuyama, R. Hayashi, K.
Otake, Y. Kurusu, Chem. Commun. 1988, 44; b) H. Kumobayashi, S.
Mitsuhashi, S. Akutagawa, S. Otsuka, Chem. Lett.1986,
157.
[5]
M. Alterman, H. O. Andersson, N. Garg, G. Ahlsén, S. Lövgren, B. Classon, U. H.
Danielsson, I. Kvarnström, L. Vrang, T. Unge, B. Samuelsson, A. Hallberg, J.
Med. Chem. 1999, 42, 3835.