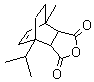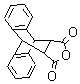[E010]
Microwave Assisted Efficient Conversion of Anhydrides to Cyclic Imides and N- Methoxy Imides.
Yousef M. Hijji* and Ellis Benjamin
Chemistry Department, Morgan State University, Baltimore, MD 21251.USA.
Abstract:
A number of cyclic imides were synthesized using cyclic anhydrides, ammonium chloride (NH4Cl), and 4-N,N-Dimethylaminopyridine (DMAP) (20% catalytic amount) catalyzed by microwave irradiation and isolated in 69 90 percent yield. A second non-N-substituted cyclic imides synthesis using cyclic anhydrides and ammonium acetate (NH4Ac) was also able to isolate cyclic imides in 59 100 percent yield. Several substituted anhydrides used were synthesized efficiently by Diels-Alder reactions of maleic anhydride with 1,3-dienes under microwave conditions. A two-step one-pot synthesis of thalidomide, a drug recently found to help numerous diseases such as leprosy, HIV, and multiple forms of cancers, was accomplished in 52 percent yield using DMAP, NH4Cl, phthalic anhydride, and glutamic acid in a conventional microwave for 6 minutes 30 seconds. Also the first microwave synthesis of N-methoxyimides in 61 99 percent yield is also reported.
Introduction:
Cyclic imides and their derivatives have been found to be an important moiety in creation of novel medicinal[1], polymeric[2], photonic[3],[4], and electronic materials[5]. Often, these imides are oxidatively stable[6], heat retardant[7], solvent resistant[8], and physically strong[9]. The conventional syntheses of substituted cyclic imides are well documented in the literature[10],[11], however the syntheses of unsubstituted cyclic imides are often limited due to the harsh conditions necessary[12],[13]. There are several conventional synthetic techniques of unsubstituted imides common used in the literature. These conditions include the condensation of liquid and/or gas ammonia with generic cyclic anhydrides[14], the cyclicization of an amide - acid with 1,1-carbonyldiimidazole (CDI) and DMAP[15], the reaction of diacid chlorides with lithium nitride[16], the reaction of a primary and a secondary amide reacted with AlCl3[17], and the reaction of urea/thiourea with a cyclic anhydride15. These conditions can often cause low yields, by-product formation and long reaction times. 13
The incorporation of microwave technology into many organic reactions has been found to increased reaction yields, decreased reaction times (1000x), and the ability to react under solventless conditions[18]. The ability to incorporate a novel microwave synthesis for the high yield production unsubstituted cyclic imides of desired material is of interest. Specifically the understanding of the microwave synthesis of unsubstituted cyclic imides is an important step forward in of how to create low-cost, high yield cyclic imides and its derivatives. Seijas et. al.[19] were able to incorporate microwave radiation with urea / thiourea and generic cyclic anhydrides to find 85 percent yield. Repetition of this work found similar result while incurring high pressure and the development of a malodorous smell. In view of the importance of this functionality, and the development of microwave application in organic synthesis, it is surprising that few reports have appeared addressing expeditious synthesis of thalidomide and cyclic imides.
Discussion:
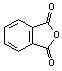
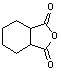
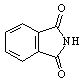
We wish to report two novel methods of synthesizing non-N-substituted cyclic imides. The first method uses a cyclic anhydride, ammonium chloride, and DMAP under microwave heating as an efficient practical one step synthesis, See scheme 1-1. Anhydrides listed in table 1 (a) were reacted with a mixture of ammonium chloride and 10% mole equivalents of DMAP. The mixture was heated in a commercial kitchen microwave[20] for 3-4 minutes. The mixture melted to a brown liquid. The heating continued for an additional one minute then allowed to cool. The products were obtained by flash chromatography, purified further by recrystallization. The yields were 80-92% table 1(a). A second method using ammonium acetate was also found to be similarly efficient as our first method, See scheme 1- 2. Anhydrides listed in table 1 (b,c) were mixed in a 1:1 ration of ammonium acetate and cyclic anhydride. This was allowed to heat in both a conventional microwave (b) and a Personal Chemistry[21] (c) for 22 sec 5 min. The mixture melted to a clear liquid, gas was evolved, and the heating continued for an additional 30 seconds then cooled to room temperature. The products were obtained by flash chromatography, purified further by recrystallization. The yields were 43-100% table 1(b,c).Monitoring the temperature of the reaction in a mono mode microwave showed that the temperature of the mixture rises very slowly beyond room temperature to about 60 oC and suddenly rises to more than 250 oC when the mixture melts.
The addition of complex anhydride systems were created through a diels-alder reaction of maleic anhydride and reactive 1,3-dienes, (anthracene, a-terpinene, and cyclohexadiene) see Table 2. Maleic anhydride and the reactive diene were mixed in a 1:1 ratio in soventless conditions and heated in a conventional microwave between 5 10 minutes. The percent yield was 92-95 providing a variety of substituted succinic anhydrides. The stereochemistry was high as the endo:exo ratios were more than 12:1. The results are presented in table 2.
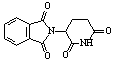
A two-step one-pot microwave synthesis of thalidomide using DMAP, ammonium chloride, phthalic anhydride, and glutamic acid in a conventional microwave for 6 minutes 30 seconds was isolated in 52 percent yield, See Table 1-11. Phthalic Anhydride, glutamic acid, and ammonium chloride were mixed ~1:1:1 ratio with DMAP (20% catalytic) and heated in a conventional microwave for 6 min 30 seconds. The mixture melted to a brown liquid. The heating continued for an additional one minute then allowed to cool. The products were obtained by solubization and concentration in ethyl acetate, and precipitation with hexanes, further purified by recrystallization.
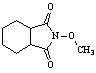
We would like to report the first microwave synthesis of N-methoxyimides in between 61 99 percent yield, See Table 3. Cyclic anhydrides and methoxyamine (HCl) were mixed in a 1:1 ratio in a conventional microwave between 17 47 seconds. The material melt in clear liquid, gas was evolved, and the heating continued for an additional 5 seconds then cooled to room temperature. The products were obtained by washing with ethyl acetate and further purified by recrystallization.
Scheme 1: Reaction of anhydrides with 1) ammonium chloride, DMAP and 2) ammonium acetate under microwave heating:
Scheme 2: Diels-Alder reaction of 1,3-diene with maleic anhydride.

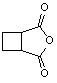
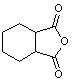
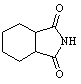
Table 1: Synthesis of non-N-substituted cyclic imides.[22]
|
Anhydride (1) |
Imide(2) |
Time (min) |
Percent Yield |
|
1) |
|
a) 5 min
b) 38 sec
c) 5min |
a) 88%
b) 73%
c) 43% |
|
2) |
|
a) 5 min
b) 47 sec
c) 5min |
a) 92 %
b) 50%
c) 81% |
|
3) |
|
a) 59 sec
b) 22 sec
c) 5min |
a) 90%
b) 92%
c) 53% |
|
4) |
|
a) 1.10 min |
a) 81%
|
|
5) |
|
a) 2 min
b) 35 sec
c) 5min |
a) 84%
b) 100%
c) 51% |
|
6) |
|
b) 47 sec 100 %
c) 5min 200 oC |
b) 88%*
c) 81%* |
|
7) |
|
a) 5 min |
a) 85% |
|
8) |
|
a) 5 min |
a) 90% |
|
9) |
|
a) 2 min |
a) dark unidentified product |
|
10) |
|
a) 4 min |
a) 80% |
|
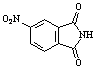
|
|
a) 3.5 min |
a) 52 % |
a) DMAP, NH4Cl (Conventional Microwave)
b) NH4Ac (Conventional Microwave)
c) NH4Ac (Personal Chemistry)
* Mixture of Nitro and Amino (Reduction) Product
Table 2: Products of Diels-Alder reaction with maleic anhydride under microwave heating.
|
Diene (3) |
Anhydride (4) |
Time (min) |
Yield (%) |
Endo / Exo |
|
1) |
|
5 min |
92% |
12 / 1 |
|
2) |
|
10 min |
95% |
15 / 1 |
|
3) |
|
10 min |
90% |
NA |
Table 3: Microwave Synthesis of N-methoxyimides.[23]
|
Anhydride(1) |
N-methoxyimide(2) |
Time |
Yield (%) |
|
|
d) 41 sec |
d) 72 % |
|
|
2) |
|
d) 17 sec |
d) 99 % |
|
3) |
|
d) 28 sec |
d) 61 % |
d) NH2OCH3-HCl (Conventional Microwave)
Acknowledgements: We would like to thank NIH for support through Research Careers in Minorities Institutions (RCMI) grant #52519.
[1] (a) Bauer, Jorg; Rademann, Jorg; Tetrahedron Letters 2003, 44 (27), 5019-5023. (b) Da Settimo, A.; Primofiore, G.; Da Settimo, F.; Simorini, F.; LaMotta, C.; Martinelli, A.; Boldrini, E. Eur. J. Med. Chem. 1996, 31,49-58.
[2] (a) Mallakpour, S. E.; Hajipour, A.-R.; Habibi, S ; European Polymer Journal 2001, 37(12), 2435-2442 (b) Stevens, M. P. J. Polym. Sci.; Polym. Lett. Ed. 1984, 22, 467-471. (c) Aitken, R. A.; Cooper, H. R.; Mehrotra, A. P. J. Chem. Soc.; Perkin Trans. 1 1996, 475-483.
[3] Mallakpour, Shadpour E.; Hajipour, Abdol-Reza; Faghihi, Khalil; Foroughifar, Naser; Bagheri, Javad. Journal of Applied Polymer Science 2001, 80(13), 2416-2421.
[4] E Mallakpour, Shadpour; Hajipour, Abdol-Reza; Faghihi, Khalil. Polymer International 2000, 49(11), 1383-1388.
[5] Struijk, Corien W.; Sieval, Alexander B.; Dakhorst, Jarno E. J.; van Dijk, Marinus; Kimkes, Peter; Koehorst, Rob B. M.; Donker, Harry; Schaafsma, Tjeerd J.; Picken, Stephen J.; van de Craats, Anick M.; Warman, John M.; Zuilhof, Han; Sudholter, Ernst J. R.; J. Am. Chem. Soc. 2000, 122 (45), 11057-11066.
[6] Zhou, Qi-Zhong; Jiang, Xi-Kui; Shao, Xue-Bin; Chen, Guang-Ju; Jia, Mu-Xin; and Li , Zhan-Ting;, Organic Letters 2003, 5 (11), 1955-1958-1958
[7] Preparation of flame-retardant and heat-resistant isocyanurate-modified imide foams. Uchiyama, Koji; Hitai, Takao; Imai, Yoshio. Patent Application: JP 86-314703 - written in Japanese. 19861226.
[8] He, Jionghao; Horie, Kazuyuki; Yokota, Rikio; He, Feifeng Polymer 2001, 42, 4063 4072
[9] Liaw, Der-Jang ; Liaw, Been-Yang ; Polymer 2001, 42, 839845
[10] (a) Meyers, A. I.; Lefker, B. A.; Sowin, T. J.; Westrum, L. J. J. Org. Chem. 1989, 54, 4243-4246. (b) Mehta, N. B.; Philips, A. P.; Lui, F. F. Brooks, R. E. J. Org. Chem.1960, 25, 1012-1015.
[11] (a) Flaih, N.; Pham-Huy, C.; and Galons, H. Tetrahedron Lett. 1999, 40, 3697-3698. (b) Vidal, T.; Petit, A.; Loupy A.; Gedye R. N.; Tetrahedron, 2000, 56, 5473-5478. (c) Bose, A. K.; Manhas, M. S.; Ghosh, M.; Raju, V. S.; Tabei, K.; Urbanczyk-Lipkowska, Z. Heterocycles 1990, 30, 741-744. (d) Bose, A. K. Organic Synthesis, Collective Vol 5, 1973, pp 973-975.
[12] Reddy, P. Y.; Kondo, S.; Toru, T.; Ueno, Y. J. Org. Chem. 1997, 62, 2652-2654. and references therein.
[13] Garner, P. Ho, W. B.; Grandhee, S. K.; Youmg, W. S.; Kennedy, V. O. J. Org. Chem. 1991, 56, 5893-5903.
[14] Polonaski, Tadeusz ; Milewska, Maria J.; Gdaniec, Maria; Tetrahedron Asymmetry 2000, 11, 3113-3122
[15] Muller, George W. ; Konnecke, William E.; Smith, Alison M.; and Khetani, Vikram D.; Organic Process Research & Development 1999, 3, 137-140
[16] Gordon, A. J.; Ehrenkaufer, R. L. E. J. Org. Chern., Vol. 36, No. 1, 1971
[17] Bon, Eric; Reau, Regis; Bertand, Guy; Bigg, Dennis C.H.; Tetrahedron Letters, 1996, 37,8, 1217-1220.
[18] Lidstrom, Pelle; Tierney, Jason; Wathey, Benard; Westman, Jacob Tetrahedron 2001 9225-9283
[19] (a) Seijas, J. A.; Vazquez-Tato P.; Montserrat-Martinez, M.; Nunez-Corredoira G. J. Chem Res. (S) 1999, 420-421. (b) Seijas, Julio A.; Vázquez-Tato, M. Pilar; González-Bande, Cristóbal; Martínez, M. Montserrat; Pacios-López, Beatriz; Synthesis 2001, No. 7, 9991000
[20] Kennmore Microwave Oven (Household) Output: 1100 Watts (Frequency: 2450 MHz)
[21] Personal Chemistry Emrys Optimizer
[22] Typical procedure for imide synthesis: Phthalic anhydride (1.0 g, 6.75 mmol) was placed into an 8.0 mL borosilicate vial. Ammonium chloride (0.72 g, 13.5 mmole) was added, then DMAP (0.1 g, 0.82 mmol). The vial was capped and the mixture was heated to 3.5 min. in the microwave at full power. The solid melted and a clear liquid formed. The heating was continued for one additional minute and the reaction mixture was allowed to cool to room temperature. The mixture was treated with 5 ml of acetone, then flash chromatographed on 30 g silica gel using ethyl acetate as an eluent. This gave 0.92 g (92%) of phthalimide as a yellowish solid. MP 226-228. 13C NMR (90 MHz, CDCl3) d 123.0, 132.7, 134.4, 169.3; MS m/z 147 (M +); IR (chloroform) (nmax, cm-1): 1740.9, 1778.2 (2 C=O)
[23] Typical procedure for N-Methoxyimide synthesis: Phthalic Anhydride (1.0 g, 6.75 mm) and Methoxyamine (HCl) (0.56 g, 6.75 mm) were mixed in 8ml tapped glass vial. This was allowed to heat for 41 seconds then cooled to room temperature to afford a white solid. This was extracted with Ethyl Acetate isolated, dried, and rotovaped to afford a white solid 0.86 grams (72%). MP 117-120. 13C NMR (90 MHz, CDCl3) d 65.6, 123.3, 134.3; MS m/z 177 (M +); IR (chloroform) (nmax, cm-1): 1733.0, 1790.1 (2 C=O)

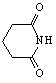

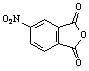


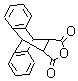
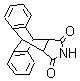

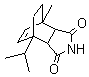
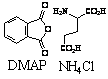 11)
11)