|
[A006] |
||
| Nucleophilic Introduction of Functional Carbon Substituents into Quinolines, Quinolones and Related Heterocycles | ||
|---|---|---|
| Wolfgang Stadlbauer*, Hana Prokopcova, Jana Sefcovikova, and A. Elisabeth Täubl | ||
|
|
Institut für Chemie,
Organic Synthesis Group, |

|
| Received: July 4, 2003/revised: Sept. 1, 2003/Uploaded: ##. #### 2003 | ||
| Contents: |
| General | |
|---|---|
|
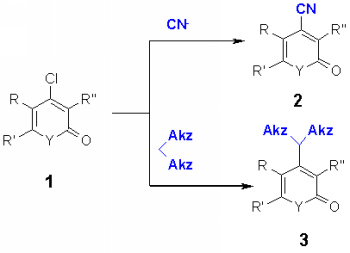
Functionalization of heterocycles by introduction of a new C-C bonding between the heterocyclic nucleus and a carbon substituent having either suitable structural properties or offering subsequent reactions is opening a broad field of new structures either with biological interest or with interesting properties e.g. in material science. Whereas C-C coupling in aromatic chemistry follows in most cases the mechanism of electrophilic substitution, in heterocyclic chemistry the nucleophilic substitution is preferred for the large field of pi-deficient heterocycles such as pyridine and its related heterocycles. In this communication we report about the introduction of cyano groups and CH-acidic compounds into quinolines and related compounds (1) by nucleophilic substitution of reactive leaving groups such as chloro groups. |
| 1. Introduction of Cyano Groups | |
|---|---|
|
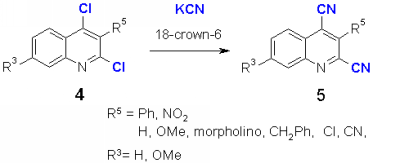
In the course of our study of nucleophilic introduction of functional groups into p-deficient heterocycles [1] we focused our interest on the introduction of cyano groups, which offer in many cases a versatile functionalization leading to carboxyl derivatives or alkylamines. The reaction of 2,4-dichloroquinoline with cyanide anion can give three possible products according to our findings [1]: exchange of the chlorine atom in either 2, or in 4-position or in both positions. The synthesis of 2-chloroquinoline-4-carbonitriles and 4-chloroquinoline-2-carbonitriles is described in the literature, however not via direct chloro exchange [2]. The reaction of 4 was carried out with potassium cyanide in DMFas solvent and 18-crown-6 as catalyst, similar to the reactions with the azide anion [1]. With electron acceptor groups R5 such as nitro and phenyl, the dicarbonitriles 5 are formed in good yields. No influence of an electron-donating substituent R3 at position 7 could be observed. It was not possible to obtain monocarbonitriles by changing the ration of potassium cyanide or the reaction conditions such as reaction time, temperature or solvent. Also the use of protic solvents which produced with azides 2-monosubstituted products [1], gave no results. When dichloroquinolines 4 with electron donating such as methoxy, benzyl, substituents R3 or with electron acceptor substituents such as cyano or chloro were reacted, the yield of 5 was much lower and a number of byproducts were obtained which made the isolation difficult.
The hydrolysis of 5 in HCl/ether did not allow to obtain the corresponding amides. After 2 days reaction time a part of the starting material remained unchanged, and 2 new compounds could be detected in the TLC. Similary, heating in conc. HCl gave a mixture of compounds.
|
|
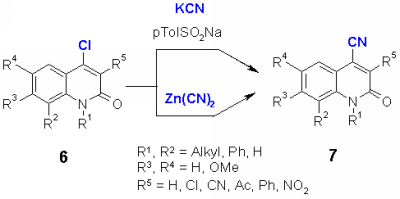
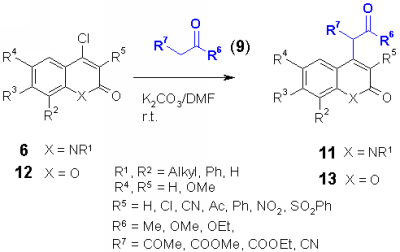
4-Chloro-N-substituted quinolones 6 did not react with cyanide anion with cyanide and crown ether. In the literature, the use p-toluenesulfinates as catalyst [3] or via heavy metal cyanides such as zinc or copper cyanides [4] was described for the cyanide introduction into chloro heterocycles. Actually, the chloroquinolones 6 reacted in good yields with potassium cyanide in dry DMF using sodium- or lithium p-toluenesulfinate as catalyst to give the nitriles 7. The reaction of 6 with zinc cyanide gave similar results, however the products 7 obtained in this reaction were more difficult to purify.
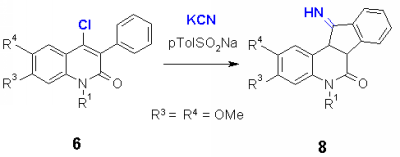
4-Chloro-3-phenylquinolones 6 having 2 methoxy substituents in the benzo part of the ring, reacted with potassium cyanide and p-toluenesulfinate to a product which showed the same mass as 7, but did not show the cyano signal in the IR spectrum at about 2240 cm-1. The structure could be assigned to the imine 8, which can be hydrolyzed to the oxo derivative. With zinc cyanide, both products, 7 and 8 were formed which could be separated chromatographically.
|
| 2. C-C Coupling with Carbanions | |
|---|---|
|
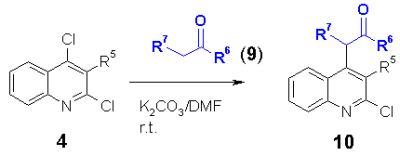
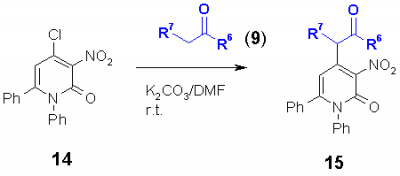
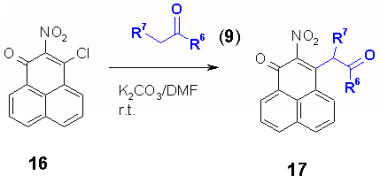
Nucleophiles such as CH acidic compounds offer a further kind of synthons which should react with chloro heterocycles in a nucleophilic substitution. The products are obviously well functionalized for further reactions. As heterocycles, 2,4-dichloroquinolines 4, 4-chloroquinolones 6, 4-chlorocoumarins 12 and 4-chloropyridones 14 were used. In addition, 3-chlorophenalenones 16 without a heterocyclic system was used.
As carbanions malonates, cyanoacetates, acetylacetonates and dimedone (9) were used. The appropriate CH acidic compound was reacted at room temperature with the corresponding chloro heterocycle in DMF using potassium carbonate as base. It was found that electron acceptor groups such as the nitro group in the ortho position of the chloro group accelerate the reaction speed enormously. Also other electron acceptor groups such as chloro, cyano, acetyl, phenyl and phenylsulfonyl increase the reactivity of the chloro group to give the corresponding heterocyclic substituted malonates, cyanoacetates, acetylacetonates and dimedons 10, 11, 13, 15 and 17 in excellent yields. |
|
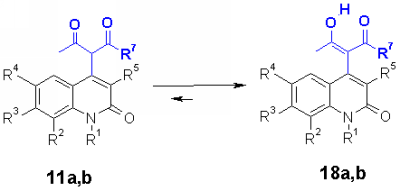
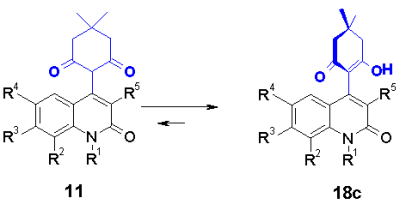
The structure elucidation revealed that acetylaconates 11a and acetoacetates 11b exist in a keto enole tautomeric equilibrium 18a,b. The cyclohexenyl ring of enolized dimedon 11c in addition shows a steric hindrance of rotation shown in 18c which can be observed by doubled NMR signals of the relevant protons.
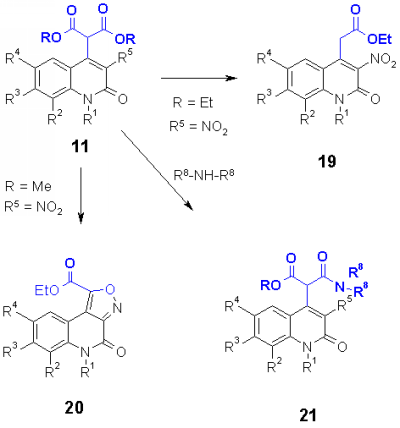
In a preliminary publication [5] we have already reported about the thermolytic degradation of heterocyclic malonates 11 which cyclized either to isoxazoles 20 or reacted to acetic acids 19 depending on the substituents [5]. Heterocyclic malonates 11 could also be reacted with primary and secondary aliphatic amines to give regioselectively heterocyclic malonamates 21.
|
| 3. Experimental |
|---|
|
General Procedure for the synthesis of quinoline-2,4-dicarbonitriles 5
General procedure for the synthesis of 2-oxo-quinolin-4-carbonitriles 7
General procedure for the synthesis of 4-substituted quinolin-2-ones (10, 11, 13,15,17,18)
|
| 4. Acknowledgement |
|---|
This work was supported by scholarships within the CEEPUS network (J.S., H.P.). |
| 5. References | |
|---|---|
|
[1] Fiala W., Stadlbauer W., Journal f. Prakt. Chem. 335 (1993), 128-134
Steinschifter W., Stadlbauer W., J. Prakt. Chem. 336 (1994) 311-318 Stadlbauer W., Hojas G., J. Chem. Soc., Perkin Trans 1, 3085 - 3087 (2000). A. E. Täubl, K. Langhans, Th. Kappe, W. Stadlbauer, J. Heterocyclic Chem., 39, 1259-1264 (2002). [2] Belli M. L., Illuminati G., Marino G., Tetrahedron 19 (1963) 345.
|
[3] Miyashita A, Suzuki Y, Ohta K, Higashino T, Heterocycles, 39 (1994) 345-55.
|