
7th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-7), http://www.mdpi.net/ecsoc-7, 1-30 November 2003
[A008]
Davor Margetiæ* and Mirjana Eckert-Maksiæ*
Laboratory for Physical Organic Chemistry,
Department of Organic Chemistry and Biochemistry,
Ruðer Bokoviæ Institute, Bijenièka c. 54, 10000 Zagreb, Croatia
E-mails: [email protected]; [email protected]
___________________________________________________________________________________________________________________________
Keywords:
organometallic dienes, norbornenes, Diels-Alder reaction, transition state, activation energy, semiempirical AM1 calculationsAbstract
. High pressure synthesis of novel policyclic structures containing germanium and silicon atoms placed at the bridgehead positions, (organometallic derivatives of bicyclo(2.2.1)hept-2-ene) is described. In addition, investigation involves semiempirical quantum-chemical study of cycloaddition properties of metalloles (Si and Ge) in Diels-Alder reactions.____________________________________________________________________________________________________________________________
Introduction.
1-sila-2,3,4,5-tetraphenyl-1,1-dimethyl-2,4-cyclopentadiene and 1-sila-2,5-diphenyl-1,1-dimethyl-2,4-cyclopentadienes (siloles) are reactive dienes, which readily undergo [4p+2p] cycloadditions with activated alkynes or alkenes to give 7-silanorbornadiene or 7-silanorbornadiene derivatives, respectively.[1] Work on cycloaddition of germoles has been also reported, [2,3,4] while stannole cycloadducts are known to be thermally unstable.[5] In recent years we have been interested in exploring cycloadditions to thermally labile 7-oxanorbornene derivatives under high pressure conditions.[6,7,8,9] This synthetic technique opened an avenue to a novel class of compounds possesing 7-sila (7-germa-) norbornene ring moiety.[10,11,12] Some of the compounds prepared in the course of these studies are shown in Figure 1.
The work reported here cover the synthesis of more complex systems starting from 1,2-dimethyl-2,3,4,5-tetraphenyl silole and 1,2-dimethyl-2,3,4,5-tetraphenyl germole as dienes and anti-1,4:5,8-diepoxy-1,4,5,8-tetrahydroanthracene as dienophile.

Results and discussion.
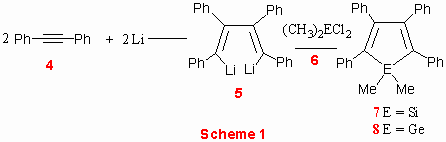
1,2-dimethyl-2,3,4,5-tetraphenyl metalloles 7[13] and 8[14] are easily accessible by reaction of lithium with diphenyl acetylene, followed by addition of corresponding dichlorodimethyl organometallic reagent (Scheme 1).
All Diels-Alder cycloaddition reactions of 1,2-dimethyl-2,3,4,5-tetraphenyl metalloles with various cyclic dienophiles were carried out under high pressure. Products and stereochemical outcomes of the reactions were analysed by NMR spectroscopy.
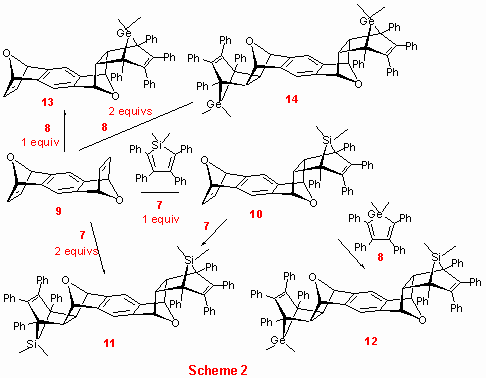
Summary of the studied reactions is shown in Scheme 2. Reaction of 1,2-dimethyl-2,3,4,5-tetraphenyl silole 7 with an equimolar amount of anti-1,4:5,8-diepoxy-1,4,5,8-tetrahydroanthracene 9,[15] gave 1:1 exo,endo- adduct 10 as a single product in 64 % yield (8 kbar, 70 oC, DCM, overnight).[16] Similarly, when the same reaction was performed in the presence of two equivalents of 7, symmetrical di-silicon adduct 11 was formed in 49 % yield. Alternatively, the adduct 11 was obtained by heating 10 with 7 (at 8 kbar, 70 oC, DCM, overnight, 71 %). In the second set of experiments reactivity of 1,2-dimethyl-2,3,4,5-tetraphenyl germole 8 under identical reaction conditions was explored. Thus, the germanium counterpart 13 of the silicon exo,endo- adduct 10 was prepared by reaction of 8 with 9 in 25 % yield. We also succeded in preparing the mixed silicon/germanium adduct 12 and di-germanium cycloadduct 14 (in 7 and 14% yield, respectively, as estimated by 1H-NMR spectroscopy).
Adducts 10-14 are among the first examples of an organometallic polynorbornene compounds containing two metal atoms at the bridgehead positions prepared in our group.[17] AM1 modelling of adduct 12 (Figure 3, phenyl substituents are omitted for sake of simplicity) has shown that spatial separation between two metal atoms is 11.8 A.
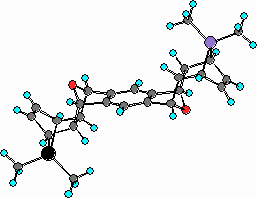
Figure 2
AM1 optimized structure of compound 12.
These results clearly show the great advantage of application of high pressure in cycloaddition reactions of the studied class of compounds. We have also examined possibility of preparing the same adducts in thermally conducted reactions (DCM, overnight or glass sealed tube at 120 oC, for 3 days). In that case, much less product was obtained accompanied with polymeric material.
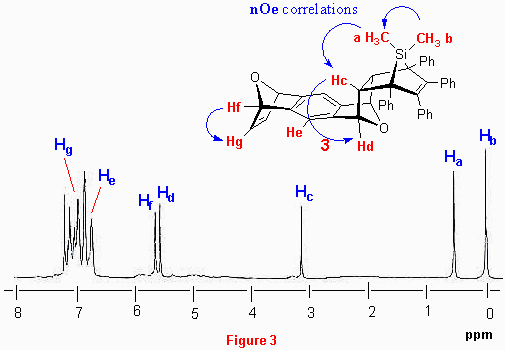
Elucidation of the stereochemistry of adducts 10-14 using 1H-NMR spectroscopy is illustrated on the example of 1:1 adduct 10. The exo,endo- structure of 10 was assigned by using standard 1D and 2D 1H-NMR spectroscopy (combining correlations obtained by 1H-1H-COSY and 1H-1H-NOESY experiments). The 1H-NMR spectrum of 10 is shown in Figure 3. All olefinic and aliphatic proton resonances occur as singlets in the spectrum, showing that molecule possess Cs symmetry. Two methyl resonances occur close to the TMS position, and are assigned to silicon substituted methyl groups at 0.03 and 0.57 ppm. Methyl group Ha syn with respect to the double bond is shielded and appears upfield compared to the shift of the analogous protons of anti methyl group Hb. Furthermore, methyl protons Hb show a nOe correlation with endo- protons Hc (d 3.17). Endo protons Hc also correlate with oxa bridgehead protons Hd at d 5.59 (as seen in the COSY and NOESY spectra). Singlet resonance at d 5.67 belongs to the second oxa bridgehead proton Hf, which correlates with olefinic bond protons Hg hidden in aromatic area. Singlet corresponding to the central aromatic proton He is also overlapping with aromatic protons (multiplet d 6.87-7.21).
One interesting feature of the spectrum is that there is no significant difference in the chemical shifts of protons Hd and Hf. On the basis of previously published data and our experience with policyclic bridged systems, we would expect that protons Hd occur at higher field (at around 5 ppm).[18] It is reasonable to assume that the observed trend is a consequence of magnetic effects of deshielding cone of aromatic substituents on silole. Important nOe correlations are depicted as blue arrows in Figure 3. Finally, we mention in passing that the 13C-NMR spectrum possesses 19 lines in total, 11 of them are aromatic and 6 aliphatic.
Part 2. Semiempirical quantum chemical study
In the second part this contribution we summarize results of RHF/AM1 calculations. Fully optimized semiempirical calculations (at the RHF/AM1 level) have been carried out in an effort to obtain additional insight into the chemistry and stereospecificity of metalloles described above. The studied compounds are displayed in Figure 4, while the calculated activation energies are collected in Tables 1-3. It should be mentioned that the AM1-calculated transition state geometries for all reactions studied are consistent with a concerted [4p+2p] cycloaddition process and only TSs with unsymmetrical dienes were found to be slightly asynchronous. An example (for reaction of silole 2 with 1,4-epoxy-1,4-dihydroanaphthalene) is shown in Figure 5.

Figure 5
AM1 optimized TS structure for reaction of silole 2 with 1,4-epoxy-1,4-dihydroanaphthalene
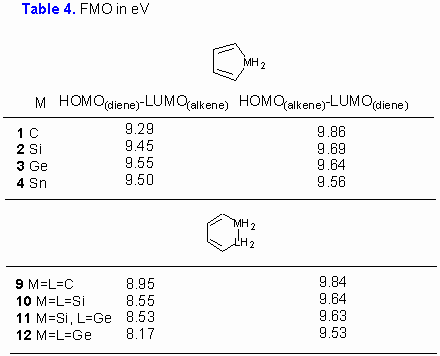
In the first set of calculations (Table 1), we have calculated the critical points for reactions of unsubstituted dienes 1-4 with 1,4-epoxy-1,4-dihydronaphthalene. Only the exo- approach to the p-bond of 1,4-epoxy-1,4-dihydronaphthalene was examined. [19] Activation barriers for addition of unsubstituted dienes 1-4 to 1,4-oxa-1,4-dihydro-naphthalene are calculated to be 134.6, 135.4, 149.3 and 157.5 kJ/mol, respectively (for energetically favored exo,endo- diene approach, Table 1). This in turn suggests that cycloaddition reactivity of germoles and stannoles should be sufficiently high, especially when cycloadditions are promoted by high pressure or microwave irradiation. Furthermore, is appears that reactivity of five-membered pentacyclic metalloles in Diels-Alder reactions decreases in order 1 (CH2) » 3 (GeH2) > 2 (SiH2) > 4 (SnH2).
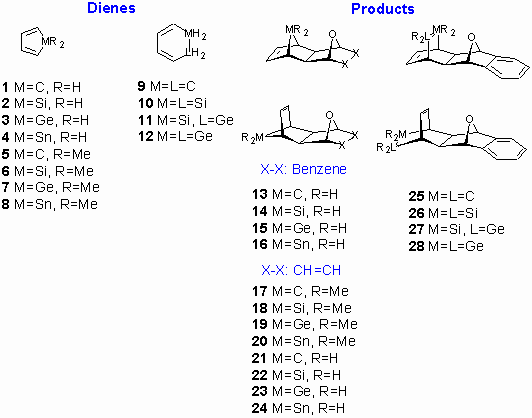
Figure 4
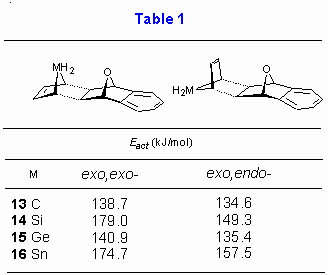
Table 2
displays the AM1 calculated activation energies for reaction of six-membered cyclic hetero-dienes 9-12. The inspection of results reveals that cyclohexadiene with two germanium atoms has the smallest activation barrier (137.9 kJ/mol), thus following the trend observed for metaloles 1-4 (Table 1). Again, the favored approach of diene is found to be from the exo,endo- side.
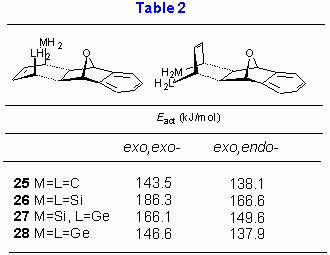
As next effect of replacement of hydrogen atoms within MH2 group with the methyl groups was explored. Reactivity of cyclic dienes 1-4 and 5-8 with 1,4-dihydro-1,4-oxanaphthalene was estimated and results are collected in Table 3. These calculations also allow an insight into the effect of replacement of the benzene ring in the dienophile component with the double bond (i.e. 1,4-epoxy-1,4-dihydronaphthalene vs. 7-oxanorbornadiene).
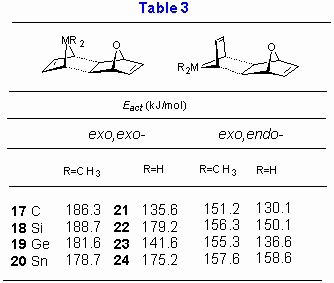
For methyl substituted dienes AM1 calculations predict larger exo,endo-/exo,exo- energy gap than for the corresponding unsubstituted metalloles (by up to 50 kJ/mol). The most plausible explanation for the observed trends lies in a more pronounced steric hindrance for the exo,exo- diene, as illustrated in Figure 6 for the exo,exo- addition of 3 to 1,4-dihydro-1,4-oxanaphthalene. It is also noteworthy that replacement of the benzene ring in 1,4-oxa-1,4-dihydro-naphthalene with double bond has no significant influence on activation energy. Furthermore it appears that methyl substitution does not alter stereochemical outcome of the reaction and the reactivity order 5 (CMe2) » 7 (GeMe2) > 6 (SiMe2) > 8 (SnMe2).
Figure 6
. TS for the exo,exo- addition of 3 to 1,4-dihydro-1,4-oxanaphthalene
Finally, it should be noted that the reactivity ordering of dienes as judged from the calculated activation energies differs from predictions based on examination of frontier molecular orbitals of metaloles (Table 4). It may be concluded that all cycloaddition reactions studied in this work are normal electron demand Diels-Alder reactions, where HOMO(diene)-LUMO(dienophile) are the dominant orbital interactions.
Computational details.
All geometry optimizations for the ground and transition states were performed using the Gaussian98[20], program package employing AM1 method, without any geometrical constraints using the Fletcher-Powell method[21] and default Gaussian convergence criteria. Transition states were verified by vibrational analysis and a possession of a single negative frequency of vibration, as illustrated in Figure 7 for reaction of dimethyl cyclopentadiene with 7-oxanorbornadiene.
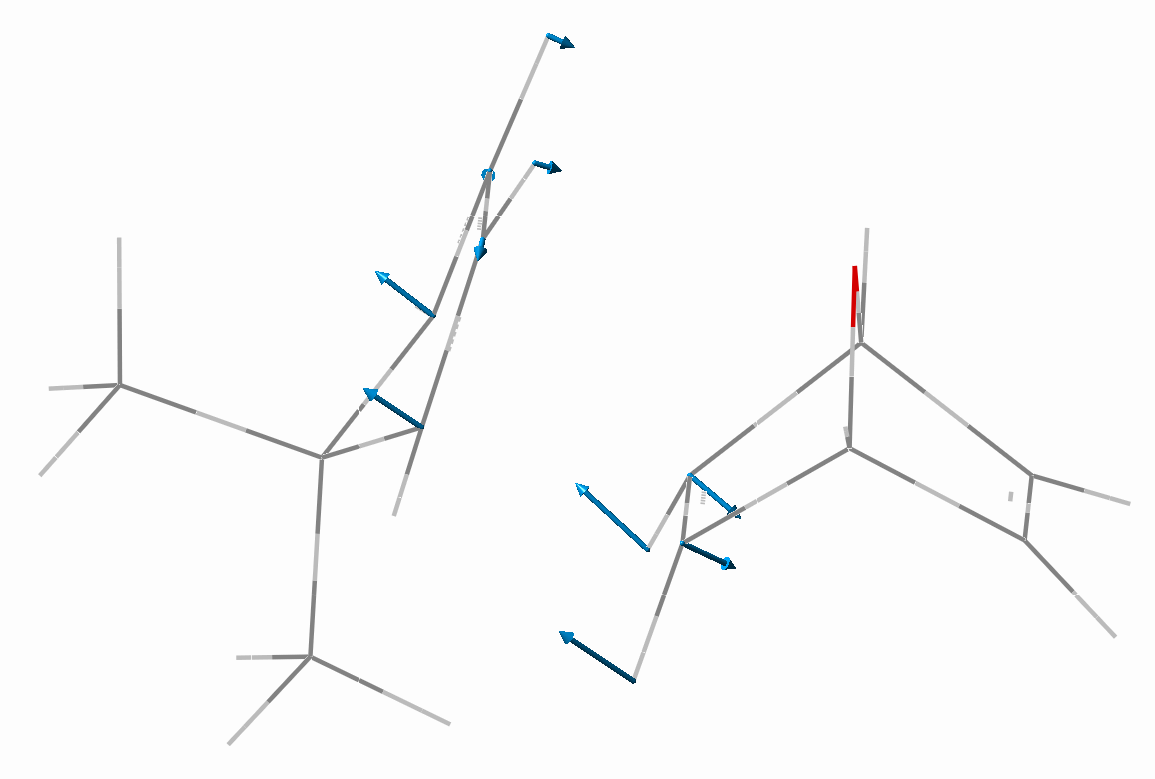
Figure 7
Conclusion.
The ability of metalloles 7 and 8 to react with anti-1,4:5,8-diepoxy-1,4,5,8-tetrahydroanthracene under high pressure conditions was explored and several novel policyclic organometallic structures prepared. Sequential addition of metalloles enables preparation of novel bis-metallic polynorbornene with rigid and well defined geometrical separation of metal centres. All studied cycloaddition reactions with metalloles exhibit high stereospecificity resulting in exclusive formation of the exo,endo- adducts. Semiempirical quantum chemical study correctly predicts experimentally observed exo,endo- stereospecificities.Acknowledgments.
Croatian Ministry of Science and Technology (MZT) is acknowledged for financial support (grant number 0098056).References.
1
. Dubac, J.; Laporterie, A.; Manuel, G. Chem. Rev. 1990, 90, 215.2
. Hota, N. K.; Willis, C. J. J. Organometal. Chem. 1968, 15, 89.3
. Scriewer, M. Ph. D. thesis, University of Dortmund, Germany, 1981.4
. Adachi, M.; Mochida, K.; Wakasa, M., Hayashi, H. Main Group Met. Chem. 1999, 22, 227; Egorov, M. P.; Ezhova, M. B.; Antipin, M. Yu.; Struchkov, Y. T. Main Group Met. Chem. 1991, 14, 19; Neumann, W. P.; Schriewer, M. Tetrahedron Lett. 1980, 21, 3273.5
. Grugel, C.; Neumann, W. P.; Schriewer, M. Angew. Chem. 1979, 91, 577.6
. Matsumoto, K.; Acheson, R. M. (eds.), Organic Synthesis at High Pressures, Wiley, New York, 1990.7
. Kirin, S. I.; Eckert-Maksiæ. M. Kem. Ind. 1999, 48, 335.8
. Margetiæ, D.; Butler, D. N.; Warrener, R. N. ARKIVOC 2002, 6, 234.9
. Margetiæ, D.; Warrener, R. N.; Butler, D. N. Aust J. Chem. 2000, 53, 959.10
. Kirin, S. I.; Vikiæ-Topiæ. D.; Mestroviæ. E.; Kaitner, B.; Eckert-Maksiæ. M. J. Organomet. Chem. 1998, 566, 85.11
. Kirin. S. I.; Klärner, F.-G.; Eckert-Maksiæ. M. Synlett 1999, 351.12
. Eckert-Maksiæ. M.; Kazaziæ. S.; Kazaziæ, S.; Kirin, S. I.; Klasinc, L.; Srziæ. D.; igon, D. Rapid Commun. Mass. Spectrom. 2001, 15, 462.13
. Ferman, J.; Kakareka, J. P.; Klooster, W. T.; Mullin, J. L.; Quattrucci, J.; Ricci, J. S.; Tracy, H. J.; Vining, W. J.; Wallace, S. Inorg. Chem. 1999, 38, 2464; Zavitoski, J. G.; Zuckerman, J. J. J. Org. Chem. 1969, 34, 4197; Gustavson, W. A.; Principe, L. M.; Min Rhe, W.-Z.; Zuckerman, J. J. J. Am. Chem. Soc. 1981, 103, 4126.14
. Mochida, K.; Wada, T.; Suzuki, K.; Hatanaka, W.; Nishiyama, Y.; Nanjo, M.; Sekine, A.; Ohashi, Y.; Sakamoto, M.; Yamamoto, A. Bull. Chem. Soc. Jpn. 2001, 74, 123; Mochida, K.; Akazawa, M.; Goto, M.; Sekine, A.; Ohashi, Y.; Nakadira, Y. Organometallics 1998, 17, 1782.15
. Hart, H.; Raju, N.; Meador, M. A.; Ward, D. L. J. Org. Chem. 1983, 48, 4357; Ashton, P. R.; Brown, G. R.; Isaacs, N. S.; Giuffrida, D., Kohnke, F. H.; Mathias, J. P.; Slawin, A. M. Z.; Smith, D. R.; Stoddart, J. F.; Williams, D. J. J. Am. Chem. Soc. 1992, 114, 6330.16
. results of corresponding syn-1,4:5,8-diepoxy-1,4,5,8-tetrahydroanthracene adducts: Kirin, S. I.; Eckert-Maksiæ, M. unpublished results17
. Kirin. S. I.; Eckert-Maksiæ. M. unpublished results18
. oxa bridged protons in coresponding exo,endo- furan adduct occur at 4.91 ppm, High-pressure facilitated cycloaddition of furan to 1,4-epoxynaphthalene, Margetiæ, D.; Warrener, R. N.; Butler, D. N., The Sixth International Electronic Conference on Synthetic Organic Chemistry (ECSOC-6), http://www.mdpi.org/ecsoc-6.htm, September 1-30, 2002.19
. endo- approach to the norbornene p-bond is higly unlikely to occur. There are rare examples in literature: Marchand, A. P.; Ganguly, B.; Malagón, C. I.; Lai, H.; Watson, W. H. Tetrahedron 2003, 59, 1763.20
. Gaussian 98, (revision A5), Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, Jr., J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Stefanov, B. B;. Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian, Inc., Pittsburgh PA, 1998.21
. Fletcher R.; Powell, M. J. D. Comput J. 1963, 6, 163.