Synthesis of 3,5-di-(4-pyridyl)-1H-1,2,4-triazole
Li-Li Liu and Guang Yang *
Department of Chemistry, Zhengzhou University, Zhengzhou, 450001, China.
*
Author to whom correspondence should be addressed. E-mail: [email protected]; Tel.: +86 371 63912869
Received: 3 August
2007
/ Accepted: 2 October 2007 / Published: 25 March 2008
Keywords: 4-amino-3,5-di-(4-pyridyl)-4H-1,2,4-triazole, 3,5-di-(4-pyridyl)-1H-1,2,4-triazole, synthesis
Introduction
4-Amino-3,5-di-(4-pyridyl)-4H-1,2,4-triazole
(1) and 3,5-di-(4-pyridyl)-1H-1,2,4-triazole (2) are potential multi-topic bridging ligands in preparation of coordination
compounds of 1 – 3D structures.[1] In an early literature, a method to prepare
compound 1 has been reported [2],
which involves the reaction of isonicotinonitrile with excess amount of
hydrazine in ethylene glycol at 130°C. Polymeric copper complexes of 2 have been prepared by Chen et al.; 2 was generated by an in situ
cycloaddition of 4-pyridylnitrile and ammonia in presence of Cu2+
under hydrothermal condition.[3] However, no synthetic procedures about the
compound 2 – the deaminated product
of 1 has appeared in the literature
to the best of our knowledge. In this paper, we describe a facile method to
prepare compound 2 starting from
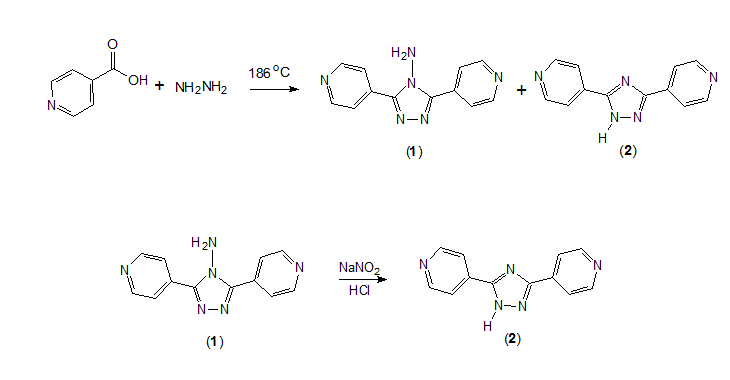
isonicotinic acid and hydrazine.
Synthesis
Isonicotinic
acid (2.46g,0.02mol)
and 2ml of 80% hydrazine were mixed in a 15 ml Teflon-lined reactor and heated for
48 h at 186°C [4]. The resulting liquid was diluted by water, and
then the pH was adjusted from 9 to 6 by addition of HCl solution. White precipitate was obtained, and recrystallised
from ethanol.
The product is actually a mixture of 4-amino-3,5-di-(4-pyridyl)-4H-1,2,4-triazole (1) and 3,5-di-(4-pyridyl)-1H-1,2,4-triazole
(2) in ca 1:7 molar ratio, estimated from the HNMR spectrum.
Pure sample
of 2 was prepared by a successive
deamination process [5]. The mixture of 1
and 2 (2.38 g,
ca 10 mmol) was dissolved in aqueous HCl
(6M, 3 mL). To it was added
slowly an aqueous solution of NaNO2 (0.76 g, 11 mmol); a lot of bubbles were generated
quickly. The mixture was stirred for 5~6 h, then the pH was adjusted from 4 to
6.5 by 10% NaOH solution. White precipitate appeared and it was filtered and
dried in air. Recrystallisation of the crude product from ethanol afforded colorless
needles
of 2. The overall yield of this
method is 9%.
Melting point: sublimated at 242°C
IR (KBr pellet, cm-1): 1607 s, 1448 m, 1144 w, 983 m, 838 w, 724 m,
513 w.
1H-NMR
(400 MHz, DMSO-d6): δ = 8.78 (4H, d, py-H), 8.02 (4H, d, py-H).
Elemental Analysis: Calculated for C12H9N5·0.5H2O: C 57.59, H 4.83, N 27.98%; Found: C 57.28, H 4.70, N 27.90%.
MS (m/z):
225.2 (15%), 224.2 (100%, M+H), 218.3 (5%).
References
1. Dong,
Y.-B.; Wang, H.-Y.; Ma, J.-P.; Huang, R.-Q.; Smith, M. D. Cryst. Growth Des.
2005, 5, 789 – 800.
2. Bentiss, F.;
Lagrenée, M.; Traisnel, M.; Mernari, B.; Elattari, H. J. Heterocyclic Chem. 1999, 36, 149 – 152.
3. Zhang, J.-P.; Lin, Y.-Y.; Huang, X.-C.; Chen, X.-M. J. Am. Chem. Soc. 2005, 127, 5495 – 5506.
4. Herbst, R. M.; Garrison, J. A. J. Org. Chem. 1953,18,872 – 877.
5. Mosch-Zanetti, N. C.; Ferbinteanu M.; Magull,
J. Eur. J. Inorg. Chem. 2002, 950 – 956.©
2008 by MDPI (http://www.mdpi.org/).
Reproduction is permitted for noncommercial purposes.