
Tetrazole binding to amidine bases.
Lars Peters, Roland Fröhlich, Alan S. F. Boyd, and Arno Kraft*
Department of Chemistry, Heriot-Watt University, Edinburgh EH14 4AS, United Kingdom.
Tel. +44 (0)131 4518040, Fax +44 (0)131 4513180, E-mail: [email protected], http:///
Received: 30 July 2001 / Uploaded 22 August 2001
Abstract: The binding of a tetrazole to an N,N'-diethylsubstituted benzamidine has been studied by X-ray crystallography and solution NMR methods. The amidinium group of a model complex was found to prefer an (E,Z) configuration in the crystal. Surprisingly, the binding mode was different in neat CDCl3, and it changed with concentration. At low concentrations, the amidine group in the complex adopted an (E,E) configuration as it does in all carboxylic acid-amidine complexes, whereas at higher concentrations the (E,Z) isomer predominates. Tetrazole turns out to be a highly flexible ligand that can easily adapt to different binding modes. This is likely to be important not only in supramolecular complexes, but also in the binding of tetrazole-containing drugs at their in vivo receptor binding sites.
Keywords: Supramolecular Chemistry, hydrogen bonding, tetrazole, amidine.
Introduction
Non-covalent binding interactions play an important role in Nature. Proteins rely on hydrogen bonding for their secondary and tertiary structure, the self-assembly of enzyme clusters (even entire viruses), and the molecular recognition of hormones and synthetic drugs at receptor binding sites. The salt bridge between a carboxylate and arginine's guanidinium group is another prominent structural motif. In modern drug design, a guanidine is often replaced by an amidine, which works quite well since amidines and guanidines are organic bases with comparable basicity.
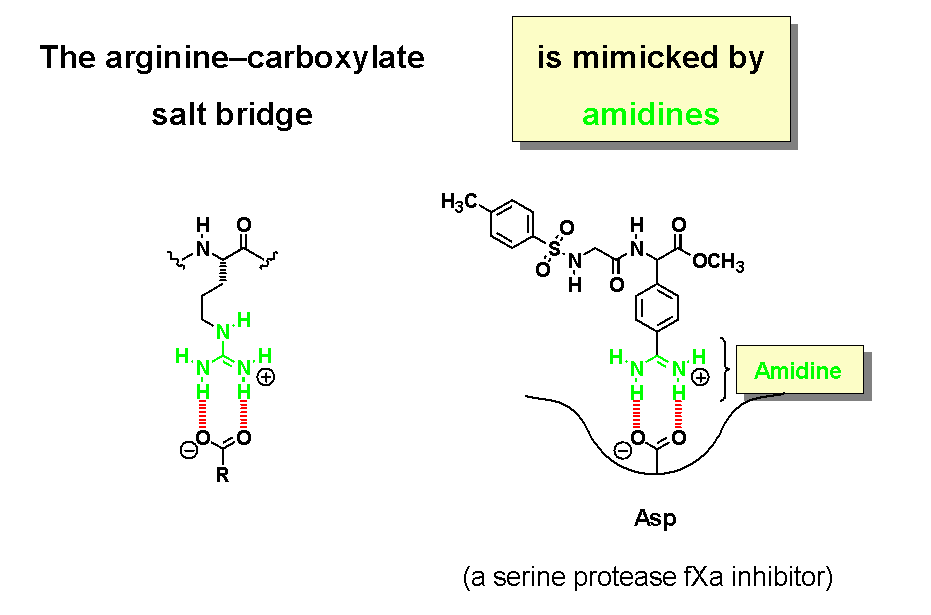
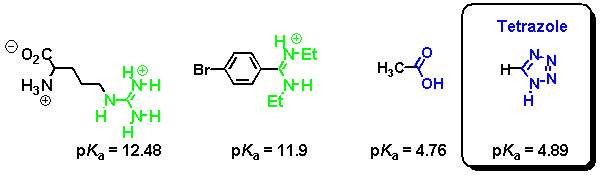
In a similar manner, tetrazoles are acidic heterocycles that serve as bioisosteric replacements for carboxylic acids. Typical pKa values are shown underneath.
The tetrazole pharmacophore has been used extensively during the past two decades in the development of angiotensin II receptor antagonists, a class of antihypertensive drugs.[1] Under physiological conditions, tetrazoles are deprotonated. According to mutagenesis studies, Losartan's tetrazolate anion binds to a protonated lysine and a histidine at the receptor binding site.[2] An X-ray crystal structure that provides further enlightenment on the actual binding pocket in the transmembrane receptor has so far not been obtained. In this context, a recently published crystal structure of an inhibitor of HIV-1 integrase illustrates clearly a preference of the tetrazolate to form hydrogen bonds with the amino groups of two lysines.[3]
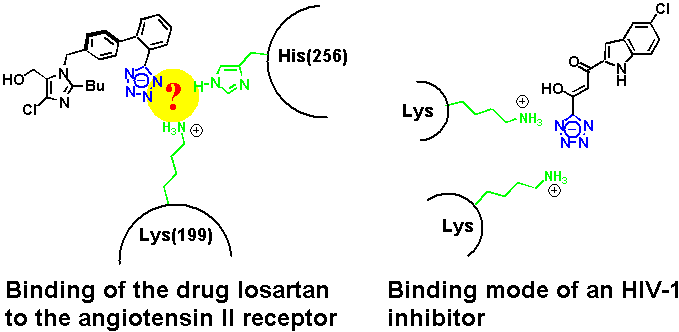
At about the same time, we reported the crystal structure of a complex between tetrazole and a heterocyclic amidine base (an imidazoline) that showed a binding mode reminiscent of the tetrazole inhibitor of the HIV-1 integrase.[4] Again, the nitrogens on one side of the heterocyclic ring are involved in hydrogen bonding to the cationic H-bond donor site.
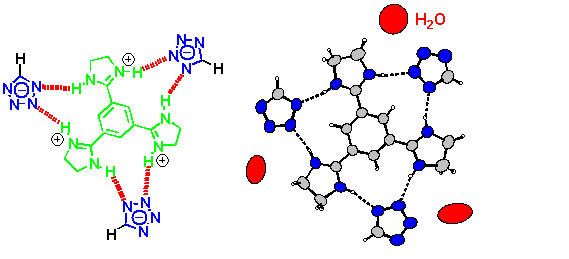
There is no evidence that tetrazolates have any tendency at all to bind to arginine, let alone to a guanidine or amidine (which are simple models for arginine) other than the above-mentioned imidazoline complex. Unfavourable electrostatic interactions have been made responsible for this,[5] and yet we wondered whether or not structural reasons may be just as important. We have therefore decided to study the binding between the parent tetrazole and a model benzamidine base.
Results and Discussion
· Initial 1H NMR dilution studies for benzamidinium tetrazolate in d6-DMSO at 25 °C show that the association constant Ka between the two ions is indeed weaker (by about 2 orders of magnitude) than for known benzamidinium carboxylates (Ka ~ 2× 103 M-1).[6,7]
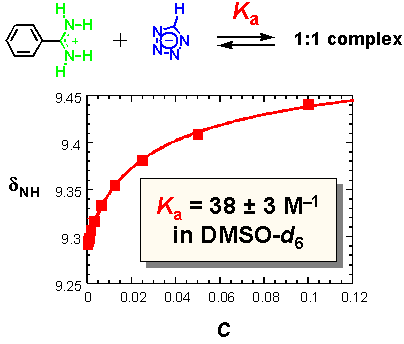
· Solution and crystal structure studies were subsequently carried out with an N,N'-diethyl-substituted amidine.
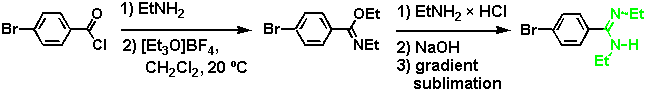
The additional ethyl groups at the amidine site allowed the complex to be dissolved in a less polar solvent, such as chloroform (where Ka was expected to be large), and different amidine isomers could be easily differentiated by NMR. This becomes again important in the determination of the solution binding structure. An N,N'-diethylsubstituted benzamidine, similar to the one chosen for the study described in this paper, has recently been introduced by Wulff et al. as the crucial binding group (and base) in molecularly imprinted polymers with esterase-like catalytic activity.[8] Compared with its parent benzamidine analogue, such an N,N'-disubstituted amidine is more soluble in nonpolar organic solvents, it has a higher stability towards hydrolysis, and aggregation is suppressed. The substituted amidine derivative offers advantages in binding studies, particularly in non-polar solvents or if weak binding interactions are expected.
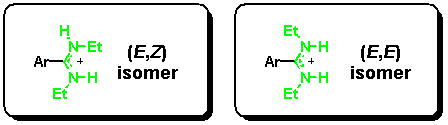
For synthetic details of the preparation of our model system, see [7]. An N,N'-diethyl-substituted amidine can exist in two configurations with an (E,E) or an (E,Z) stereochemistry at the partial C-N double bond, whereas a (Z,Z) isomer is not possible for steric reasons.[9] Although this seems to be a complication at a first glance, the different isomers can be easily differentiated by NMR, and-by serving as an NMR marker-provide important information about binding modes in solution.
· The crystal structure of a 1:1 complex between benzoic acid and the substituted amidine shows two almost linear hydrogen bonds. The binding mode is similar to that of unsubstituted amidines. As a consequence, the substituted amidine must, of course, be (E,E)-configured.
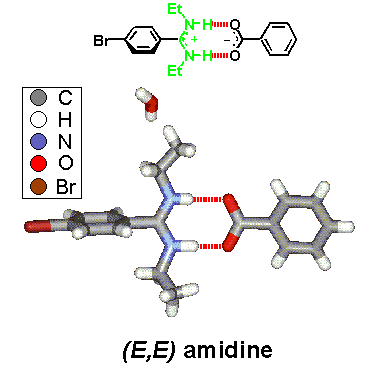
· An X-ray crystal structure of the amidinium tetrazolate shows an (E,Z)-configured amidine instead. The tetrazolate still forms two hydrogen bonds but to different amidinium ions. Hydrogens were found at the nitrogen atoms from difference Fourier calculations, proving a salt-like structure in the solid.
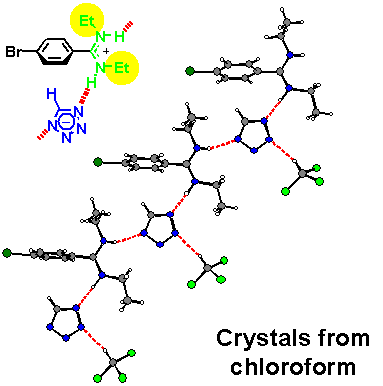
· In methanol, the amidinium tetrazolate is completely dissociated into its ionic components. Two sets of 1H NMR signals for the ethyl substituents indicate that the amidine exists exclusively as the (E,Z) isomer.
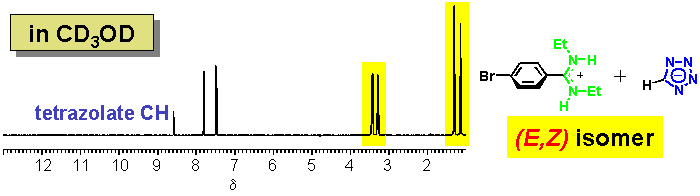
· The (E,Z) amidine is once more preferred in CDCl3 in the presence of a non-coordinating counter-anion, although additional signals can be attributed to a small amount of the (E,E) isomer.
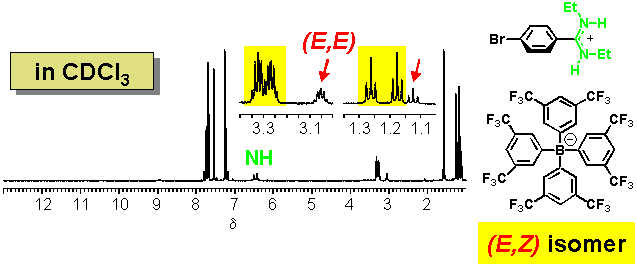
· In CDCl3, the amidinium tetrazolate is a strongly bound non-covalent complex. The downfield shift of the NH signal compared to the non-coordinating borate confirms that the tetrazolate is hydrogen-bonded to the amidine. The two ethyl groups give rise to a single set of 1H NMR signals (one triplet and one quartet). The amidine therefore prefers now to be (E,E)-configured.
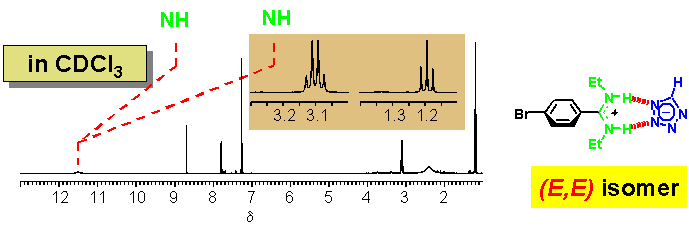
· Proton transfer from the tetrazole to the amidine-even in chloroform-is evident for the following reasons: (i) the free amidine base gives rise to broadened N-ethyl and Ar-H signals in the 1H NMR spectrum owing to extensive tautomerism and dynamic exchange between various rotational isomers; (ii) 13C and 15N NMR chemical shifts of the tetrazole are typical for a tetrazolate.
· The 1H NMR spectra of the amidinium tetrazolate in CDCl3 are concentration-dependent. With increasing concentration signals of the less symmetrical (E,Z) amidine isomer emerge. A carboxylate, in contrast, would bind exclusively to the (E,E) amidine independent of concentration.
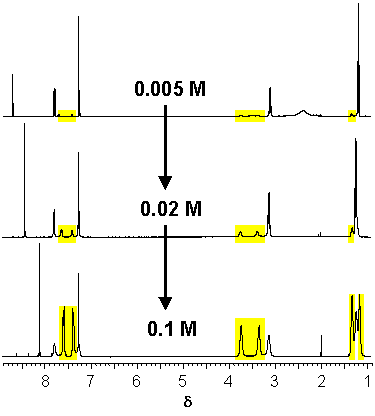
· The fraction of the (E,Z) isomer actually increases with increasing concentration of the amidinium tetrazolate in CDCl3.
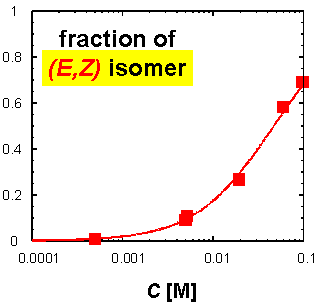
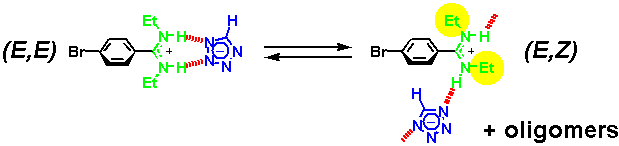
· In addition, the two amidine isomers exchange on the NMR timescale, requiring the tetrazolate to change its binding mode. The driving force behind it is probably the formation of oligomers with at least one linear (that is, strong) hydrogen bond per repeat unit. Note that the tetrazolate is smaller than a carboxylate ligand, and binding to the (E,E) amidine is therefore not ideal.
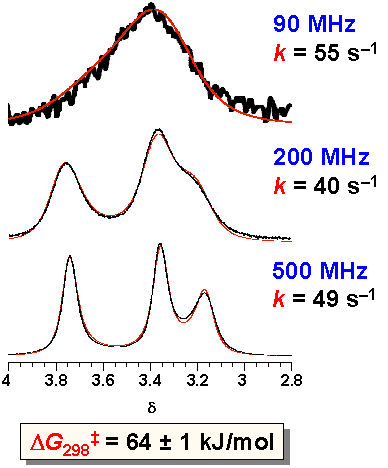
· Although it is customary in dynamic NMR spectroscopy to vary the temperature, this is not feasible in the case of our supramolecular complex since a change in temperature will invariably affect both Ka and the isomer ratio. We made therefore use of the fact that the 1H NMR spectrum of a dynamic system is not only dependent on temperature but also on the magnetic field. A free enthalpy of activation DG of 64 kJ/mol can be deduced from lineshape analyses of the same sample at the same temperature and concentration, yet at different spectrometer frequencies. The value of the rotational barrier is similar to that of other amidines, and about 20 kJ/mol less than that observed in typical amides.
Conclusions
· Tetrazoles are quite suitable ligands and can serve as replacements for carboxylic acids not only in Medicinal Chemistry, but also in Supramolecular Chemistry.
· Tetrazoles form non-covalent complexes with amidines, although there are certain pecularities.
· Most importantly, tetrazoles are highly flexible ligands and can adapt easily to different binding modes. This is likely to be important not only in supramolecular complexes, but also in the binding of tetrazole-containing drugs at their in vivo receptor binding sites.
References and Notes
[1] Wexler, R. R.; Greenlee, W. J.; Irvin, J. D.; Goldberg, M. R.; Prendergast, K.; Smith, R. D.; Timmermans, P. B. M. W. M. J. Med. Chem. 1996, 39, 625-656.
[2] Noda, K.; Saad, Y.; Kinoshita, A.; Boyle, T. P.; Graham, R. M.; Husain, A.; Karnik, S. S. J. Biol. Chem. 1995, 270, 2284-2289.
[3] Goldgur, Y.; Craigie, R.; Cohen, G. H.; Fujiwara, T.; Yoshinaga, T.; Fujishita, T.; Sugimoto, H.; Endo, T.; Murai, H.; Davies, D. R. Proc. Nat. Acad. Sci. USA 1999, 96, 13040-13043.
[4] Kraft, A.; Osterod, F.; Fröhlich, R. J. Org. Chem. 1999, 64, 6425-6433.
[5] Zablocki, J. A.; Miyano, M.; Rao, S. N.; Panzer-Knodle, S.; Nicholson, N.; Feigen, L. J. Med. Chem. 1992, 35, 4914-4917.
[6] (a) Deng, Y.; Roberts, J. A.; Peng, S.-M.; Chang, C. K.; Nocera, D. G. Angew. Chem. Int. Ed. Engl. 1997, 36, 2124-2127. (b) Papoutsakis, D.; Kirby, J. P.; Jackson, J. E.; Nocera, D. G. Chem. Eur. J. 1999, 5, 1474-1480.
[7] Peters, L.; Fröhlich, R.; Boyd, A. S. F.; Kraft, A. J. Org. Chem. 2001, 66, 3291-3298.
[8] (a) Wulff, G.; Groß, T.; Schönfeld, R. Angew. Chem., Int. Ed. Engl. 1997, 36, 1962-1964. (b) Strikovsky, A. G.; Kasper, D.; Grün, M.; Green, B. S.; Hradil, J.; Wulff, G. J. Am. Chem. Soc. 2000, 122, 6295-6296.
[9] Hammond, G. S.; Neuman, R. C. J. Phys. Chem. 1963, 67, 1655-1659.