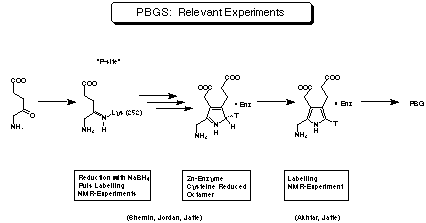
 Figure 17:
Experiments relevant for the mechanism of porphobilinogen synthase.
Figure 17:
Experiments relevant for the mechanism of porphobilinogen synthase. Mechanistic studies of porphobilinogen synthase
Proposals for the mechanism of porphobilinogen synthase
X-ray structures of porphobilinogen synthase
Mechanistic studies of porphobilinogen synthase The step leading to
porphobilinogen (4) in the biosynthesis of the "pigments of life" has been
intensively studied during the last thirty years.[38] Before the report on the first high
resolution X-ray structures of the enzyme porphobilinogen synthase appeared in the
literature end of 1997[39] a series of important experimental findings relevant for the
understanding of the enzyme mechanism have been reported (see Figure 17).[40] Almost sixty
gene derived protein sequences for porphobilinogen synthase from different sources are
known.[41] The gene derived protein sequences show a relatively high degree of homology.
The amino acids at the active site and at the postulated sites for the binding of the
metal ions which are necessary for the activity are highly conserved. Figure 17:
Experiments relevant for the mechanism of porphobilinogen synthase.
The results reported by the groups of Shemin, Jordan and Jaffe can be summarised as follows: The enzyme is usually a homo-octamer. The minimal active species is a dimer. Most of the enzymes isolated so far need Zn2+ or Mg2+ as essential cofactors. The enzymes can be categorised as a function of the metal ion needed for their activity. For those enzymes binding Zn2+ the cysteines have to be reduced to keep the enzyme in its active form. The substrate forming the propionic acid side chain is interacting first. At least one of the substrates is forming a covalent bond with the enzyme via a Schiff base. To study the order in which the two substrate molecules bind to the enzyme, Jordan performed highly elegant single-turnover experiments.[42] Stoechiometric equivalents of labelled substrate and porphobilinogen synthase were rapidly mixed and after about 100 ms added to a large excess of unlabelled substrate. The position of the radioactive label was determined by degradation. The pulse labelling could also be done using [5-13C] 5-aminolevulinic acid. The 13C-NMR spectrum of the product allowed to identify the position of the label directly.[26] The deprotonation leading from the pyrrolenine tautomer to the aromatic pyrrole is enantioselective and occurs therefore on the surface of the enzyme. This observation has been confirmed by NMR results showing that porphobilinogen (4) bound to the active site can be observed.[43] Despite the intensive efforts of several groups the exact mechanism of the biosynthesis of porphobilinogen (4) could not be deduced based on the biochemical knowledge accumulated so far.
For the detailed analysis of an enzyme mechanism the following methods are usually
applied:
1) X-ray structure determination;
2) site-directed mutagenesis;
3) spectroscopic studies;
4) kinetic and inhibition studies.[44]
Until recently most of these methods could not be applied to study porphobilinogen
synthase because no secured structural information was available. Therefore it was often
difficult to deduce clear mechanistic conclusions from the experimental results. Despite
these difficulties it was possible to analyse the sequence of the possible mechanisms
concentrating on the transformations of the substrate molecules alone.[38] This approach
provides a frame-work for the chemical analysis of the sequence of events.
Shemin has been the first to propose a mechanism for porphobilinogen synthase drawing a
close analogy between this enzyme and class I aldolases (see Figure 18).[24] Shemin was
the first to proof that porphobilinogen synthase forms a Schiff base between the e-amino
group a lysine of the active site and the carbonyl group of one of the two substrate
molecules. His postulated mechanism is mainly based on two arguments: the transformation
has been formulated in strict analogy of porphobilinogen synthase with aldolases and the
sequence of recognition is based on the observed formation of a compound which Shemin
called a mixed pyrrole.[24] Later experiments however showed that the proposed structure
was wrong.[45]
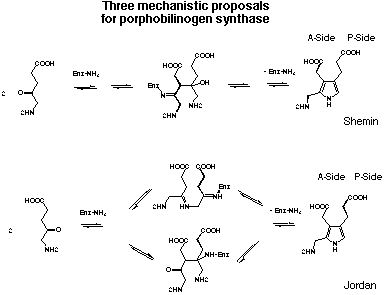
Figure 18: The three mechanistic proposals for porphobilinogen synthase.
Starting from the results of his highly elegant single turn-over experiments Jordan postulated two alternative mechanisms for the formation of porphobilinogen (4) (see Figure 18).[25,26,40] Both mechanisms include the finding that the first substrate bound to the enzyme will be incorporated into the P-side of the product porphobilinogen (4). The first alternative mechanism proposed by Jordan postulates that the first bond between the two substrate molecules is a Schiff base between the two substrate molecules. Only after this step follows the aldol reaction and the elimination which leads after deprotonation to the product. This mechanism follows closely the known mechanism for the Knorr pyrrole synthesis. In the second alternative mechanism proposed by Jordan the aldol reaction forming the carbon carbon bond is the step joining the two substrates for the first time.
Despite the efforts of several research groups, the mechanism of the enzymatic synthesis of porphobilinogen (4) is not established yet. The sequence of recognition of the two substrate molecules is: "P-site" first, "A-site" second, at least for the bovine liver and the human erythrocyte enzyme. The substrate at the "P-site" is forming a Schiff's base to a lysine of the active site. The second substrate may be bound non-covalently to the enzyme. One Zn2+ probably complexed to cysteines helps in the catalytic step. There is same circumstantial evidence, that the enzyme shows half-the-site reactivity. Assuming that the active site is formed at the interface of a dimer had been considered to be an attractive interpretation of these observations. Finally the product forms a relatively stable complex with the enzyme. This observation is in agreement with the fact that the last chemical step, the deprotonation, is still occurring at the enzyme.[46]
Since the first report on the X-ray structure determination of porphobilinogen synthase from yeast,[39] the structures of two more enzymes isolated from Escherichia coli and Pseudomonas aeruginosa have been published.[47,48] More importantly the structures of porphobilinogen synthase co-crystrallized with levulinic acid were solved as well.[47-49] The crystal structures increased our knowledge about porphobilinogen synthase considerably. Many aspects which had been inferred so far can now be clearly interpreted and correlated with the structural data. Some of the tentative conclusions, like the proposal that the active sites might be located at the interface between the monomeric units, were proven to be wrong
All structures determined are similar despite very interesting differences between the individual shapes. All three enzymes form octamers composed of dimers. The tertiary structure of all porphobilinogen synthase is dominated by a TIM-barrel.[50] The N-terminal end is not part of the TIM-barrel, but wraps around the neighbouring molecule. The N-terminal ends make major contributions to the dimer interface contacts. The eight active sites of the octamer are all exposed to the solvent. As expected for the TIM-barrel structure, the active site was found at the C-terminal end of a b-sheet. The active site lysine could be identified as well in the structure of the "empty" enzyme as well as in the structure of the co-crystals with levulinic acid. A second lysine was found to be present nearby in the active centre. This second lysine had not been identified before, but its presence seems to be crucial for the functioning of porphobilinogen synthase.[39] The yeast and Eschierichia coli enzyme contain a metal binding site near to the active site (= active site Zn) and a secondary metal binding site further removed from the active site (= structural Zn). In the structure determined for Pseudomonas aeruginosa the Mg2+-binding site is located at the surface of the subunit. The Mg2+-binding is connected to the active site via a series of hydrogen bonds, which influence the structure of the active site by a series of subtle changes. The function of Mg2+ bound to this site has been interpreted as being allosteric. Finally the active sites having bound one molecule of levulinic acid are shielded by a "lid" whereas the structures of the "empty" active sites are open towards the outside and the "lid" region is much less well defined.
The nature and the role of the basic groups at the active site can now be assessed with reasonable confidence based on the structural data.[49] The authors attribute to the second lysine at the active site a role in the formation of the Schiff base. The hydroxide ion bound to the active-site Zn is considered to be a likely candidate for the deprotonation necessary for the C-C bond formation. Finally the same Zn-ion could also co-ordinate the oxygen of the carbonyl group of A-site substrate.
One conclusion drawn from this wealth of structural information is that it is still not
possible to discriminate with certitude between the two possible mechanisms. This
uncertainty is nicely demonstrated by the title of the first publication on the X-ray
structure of porphobilinogen synthase, where the enzyme is called "a hybrid
aldolase".[39] In this and the following publication more importance is given to the
carbon-carbon bond forming process.[51,52] In the most recent publication of the same
authors, reporting on the structure of porphobilinogen synthase from Escherichia coli
complexed with levulinic acid, the conclusion drawn by the authors is the following:
"The scheme in which the C-N bond formation occurs first may be more attractive from
the mechanistic point of view since formation of the inter substrate Schiff base increases
the acidity of the A-site C-3 protons".[49]
![]()
![]() Inhibition Studies of Porphobilinogen Synthase from Escherichia
coli
Inhibition Studies of Porphobilinogen Synthase from Escherichia
coli
![]() "A Novel Synthesis of Porphobilinogen: Synthetic And Biosynthetic
Studies"
"A Novel Synthesis of Porphobilinogen: Synthetic And Biosynthetic
Studies"
Christiane Bobillier Neier / August 1999