Chemical Synthesis of Porphobilinogen
Regioselective synthesis of the silyl enol ether component
Trials to couple the silyl enol ether
Synthesis of porphobilinogen
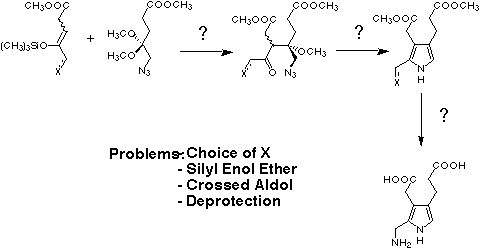
For the planned synthesis of porphobilinogen (4) and of structural analogues of porphobilinogen we needed the corresponding silyl enol ether of a protected derivative of 5-aminolevulinic acid (see Figure 13).
Figure 13: Retrosynthesis for the planned biomimetic approach to porphobilinogen (4) or to structural analogues thereof.
Trials to submit the 5-azido levulinic acid methyl ester, or the 5-tert.-butyldimethylsilyloxy-levulinic acid methyl ester to the conditions worked out by Miller[30] inevitably led to the unwanted regioisomers of the silyl enol ether. The synthetic problem could be finally solved using the 5-phthalimido-levulinic acid methyl ester 20 submitting it to Miller's conditions but changing the solvent to chloroform (see Figure 14).[31] Adding after 17 hours dry hexane allowed to precipitate the salts which had been formed. The wanted silyl enol ether 22 was obtained in 93 % yield as 1 : 1 mixture of the diastereoisomers containing only 4 % of the unwanted silyl enol ether 21 as side product.
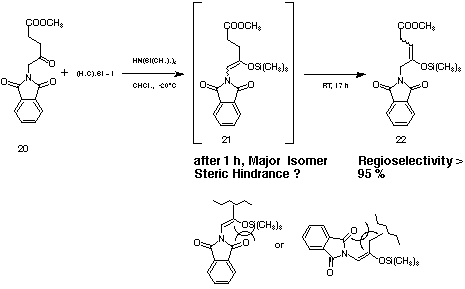
Figure 14: Synthesis of the silyl enol ether 22.
The silyl enol ether 22 could be stored for months at - 20 °C in the refrigerator. The regioselectivity of the formation of the silyl enol ether is surprising, because one has to assume the methylene group in the a-position to the phthalimido group should be clearly more acidic than the protons at the C3 methylene group. The regioselectivity can probably be attributed to the steric hindrance which is created if the silyl enol ether towards the position C3 is formed.
Having the correct regioisomer 22 in our hands experiments were undertaken to couple the silyl enol ether with the acetal of the 5-azido levulinic acid methyl ester under standard conditions.[28,32] Unfortunately all our trials to couple these two precursors of 5-amino levulinic acid according to Mukaiyama were totally unsuccessful. Despite our considerable efforts to achieve also the Mukaiyama Aldol coupling using a protected form of d-aminolevulinate, we were unable to isolate products which could be traced back to the central C-C-bond formation. Using TiCl4 as a catalyst for the aldol coupling starting from the silyl enol ether 22 at temperatures below -40 °C no reaction could be observed. Increasing the temperature above -40°C rapid destruction of the reaction partner was observed. Using Lewis acids like TMSOTf[33,34] or the "super-Lewis acid" B(OTf)4TMS according to Davis[35] the aldol reaction between 22 and the dimethyl acetal of levulinic acid methyl ester could be achieved. Using Noyori's conditions [33] whereby 0.11 equivalents of TMSOTf are utilised 30% of one pure diastereoisomer could be isolated. Even when these stronger Lewis acids were used we were unable to achieve the crucial C-C-bond forming process starting from an adequate precursor of 5-aminolevulinate. The use of the more reactive, but also more aggressive catalyst TMSI[36] at -80 °C lead to the destruction of both starting materials: the silyl enol ether 2e and the acetal.
The only way out seemed to be to increase the inherent reactivity of the carbonyl component.
In order to obtain porphobilinogen (4) we tried to use the monocyanide of succinic acid monomethyl ester (23) as activated carbonyl component. Using this strategy it should be possible to combine two partners which contain all the carbon, oxygen and nitrogen atoms necessary for the construction of porphobilinogen. Deprotection of the aldol product should then induce the ring closing and aromatisation process. In view of this analysis we reacted the silyl enol ether 22 with the monocyanide of succinic acid monomethyl ester (23). The cyano hydrine 27 could be detected in the raw product of the reaction. Extraction against water and purification with column chromatography yielded 35 % of the b-diketon rac-24 as hydrolysis product. The diketon could be easily transformed in two steps into the pyrazole 26 which is a close structural analogue of porphobilinogen (see Figure 15).[15,37]
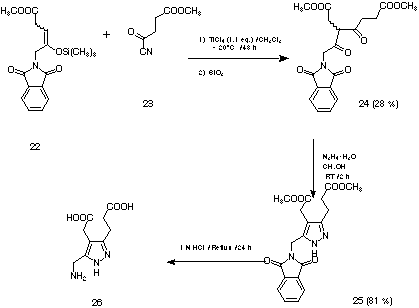
Figure 15: Synthesis of the pyrazole 26, a structural analogue of porphobilinogen.[37]
Under optimised conditions at -20° C using TiCl4, which had been freed
from HCl by distillation over polyvinyl pyridine, the aldol product rac-27 could be
obtained in 60 to 87 % (see Figure 16). One diastereoisomer of the aldol product rac-27
could be obtained analytically pure by crystallisation in 47 % yield. Trials to reduce the
cyano hydrine directly met with limited success. For the synthesis we protected the
unpurified aldol product using acetone enol acetate. The acetylated aldol product rac-28
could be obtained in 56 % yield. Even the reduction of the acetylated cyano hydrine rac-28
proved to be difficult. Finally the cyano hydrine rac-28 could be reduced smoothly
at 65° C under 120 atm H2 in the presence of Raney nickel. After column chromatography we
obtained the fully protected porphobilinogen 29[31] in 54 % yield analytically pure.
Removal of the protecting groups over two steps has already been described in the
literature.[18] 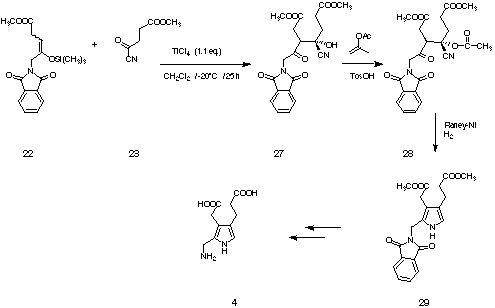
Figure 16: Synthesis of a protected form of porphobilinogen 29.
In conclusion we were able to obtain the protected porphobilinogen 29 in a convergent
way starting from two easily obtainable starting materials. The central step of the
synthesis is the Mukaiyama aldol reaction between the regioselectively formed silyl enol
ether 22 as the nucleophile with the monocyanide of succinic acid monomethyl ester (23) as
electrophile. Reducing the acetylated cyano hydrine rac-28 yields directly the
protected porphobilinogen 29. This synthesis follows the proposal for the biosynthesis
made by Shemin almost 30 years ago. The correctly functionalised side chains are
introduced on the level of the two starting materials used for the synthesis of the
pyrrole ring. Subsequent functionalisation is therefore not necessary. In this synthetic
scheme the same bonds are formed as in the biosynthesis catalysed by porphobilinogen
synthase. The overall yield starting from 5-phthalimido methyl levulinate is 25 %. The
synthesis can be used to obtain selectively labelled porphobilinogen (4).
![]()
![]() Biosynthesis of Porphobilinogen
Biosynthesis of Porphobilinogen
![]() "A Novel Synthesis of Porphobilinogen: Synthetic And Biosynthetic
Studies"
"A Novel Synthesis of Porphobilinogen: Synthetic And Biosynthetic
Studies"
Christiane Bobillier Neier / August 1999